.png)
The Bioinformatician’s Fever Dream: Data Nirvana or Just a Really Stable Manifest?
By Darren Ames, Head of Solutions Science
Filters
No Items Found!
.png)
.png)
How to Drive Innovation in Omics Data Analysis
Navigating the Complexities of Clinico-Omics Data in Precision Medicine Precision medicine stands ...
.png)
Unlocking the Potential of Precision Health in Biopharma with Advanced Data Catalogs
In today's biopharma landscape, researchers grapple with unprecedented volumes of multi-modal omics ...
.png)
Integrating Multi-Modal & Genomic and Multi-Omics Data for Precision Medicine
The Importance of Multi-Modal Data Integration in Precision Medicine Precision medicine aims to ...

DNAnexus R&D Report: Benchmarking Germline Variant Calling Pipelines - Inside DNAnexus
Variant calling is a staple of genome analysis. Developments in computational methods and ...
.png)
Accelerating External Innovation in Biopharma with Trusted Research Environments (TREs)
Discover how Trusted Research Environments (TREs) revolutionize multi-omics studies by enabling ...
.png)
Proteomics Research In Pharma: Trends, Challenges, And Recent News
Unlocking the mysteries of the proteome: How cutting-edge proteomics research is revolutionizing ...

Challenges To Scaling Genomics Pipelines In Bioinformatics Analysis
Scaling genomics pipelines in bioinformatics analysis presents unique challenges that require ...

The Challenges Of Identifying Biomarkers And Molecular Patterns: Linking Clinical And Molecular Data
Navigating the intricate landscape of biomarker discovery to unlock new therapeutic avenues in ...

Unlocking New Frontiers in Neuroscience Drug Discovery with Omics and Biomarker Discovery Breakthroughs
Unlocking New Frontiers in Neuroscience Drug Discovery with Omics and Biomarker Discovery ...

The Future of Molecular Diagnostic Testing Will Depend on TREs

Exciting Progress as New Scientists Interact with the Mexico City Prospective Study
Overview

7 Key Strategies for Addressing GxP Compliance in Biopharma
Overview

Improving Pharmaceutical R and D Productivity through Accelerated Biomarker Discovery

UK Biobank RAP Researcher Spotlight: October 2024
The Monthly Researcher Spotlight is our section highlighting the exciting work of the UK Biobank ...
Scaling Up Genomic Research: A Call to Action for Biopharma
As omics data continues to grow globally, the need for secure, collaborative data sharing has never ...
Updated NIH Genomic Data Sharing Policy: What Does It Mean for Scientists?
Author: Matt Newman Senior Vice President & General Manager, Pharma & Diagnostics DNAnexus

UK Biobank RAP Researcher Spotlight: September 2024
The Monthly Researcher Spotlight is our section highlighting the exciting work of the UK Biobank ...

UK Biobank RAP Researcher Spotlight: August 2024
The Monthly Researcher Spotlight is our section highlighting the exciting work of the UK Biobank ...

UK Biobank RAP Researcher Spotlight: July 2024
The Monthly Researcher Spotlight is our section highlighting the exciting work of the UK Biobank ...

UK Biobank RAP Researcher Spotlight: June 2024
The Monthly Researcher Spotlight is our section highlighting the exciting work of the UK Biobank ...
The Trusted Research Environment: A Gateway to Secure Collaboration
This article is the first in a series of articles about trusted research environments (TREs), and ...

UK Biobank RAP Researcher Spotlight: May 2024
The Monthly Researcher Spotlight is our section highlighting the exciting work of the UK Biobank ...
Choosing Between WDL and Nextflow for Genomics Analysis Workflows
Explore the differences between WDL and Nextflow to determine the best workflow language for your ...
Today’s Precision Medicine Requires an Enterprise Strategy
Key takeaways:

UK Biobank RAP Researcher Spotlight: April 2024
The Monthly Researcher Spotlight is our section highlighting the exciting work of the UK Biobank ...

Announcing Additional Support for the UK Biobank Research Analysis Platform
It’s been over two years since the launch of the UK Biobank Research Analysis Platform (UKB-RAP) ...

UK Biobank RAP Researcher Spotlight: March 2024
The Monthly Researcher Spotlight is our section highlighting the exciting work of the UK Biobank ...
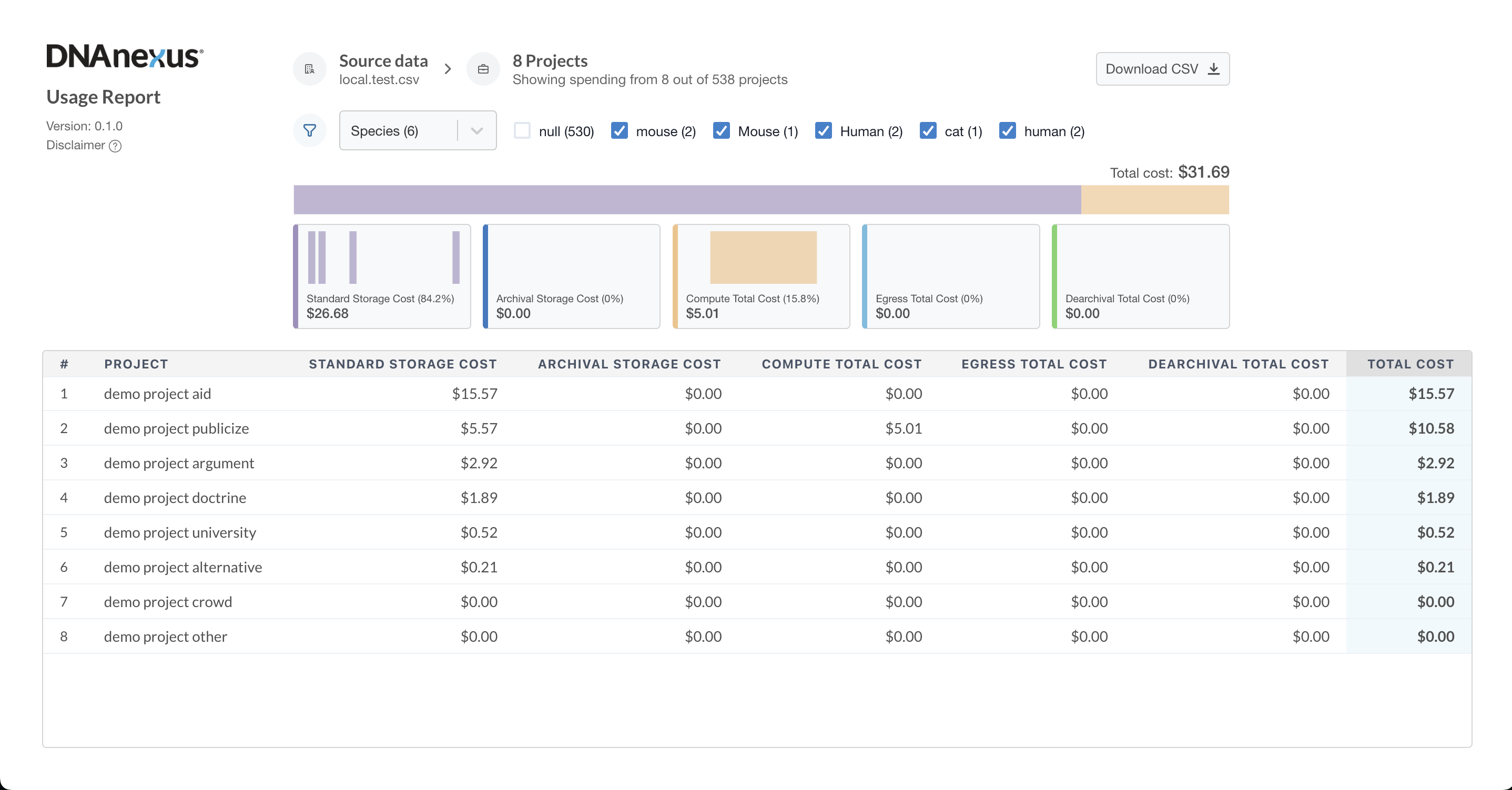
Take Charge of Your Omics Cloud Expenses
Whether you have a large user base or just a few users, managing enterprise multiomics cloud ...

From Code to Cure: Latest UK Biobank Release of Whole Genome Sequence Data from 500,000 Participants Launches New Era in Medical Research
Medical researchers around the world can now study whole genome sequencing data of half a million ...

UK Biobank RAP Researcher Spotlight: November 2023
The Monthly Researcher Spotlight is our section highlighting the exciting work of the UK Biobank ...

UK Biobank RAP Researcher Spotlight: October 2023
The Monthly Researcher Spotlight is our section highlighting the exciting work of the UK Biobank ...

UK Biobank RAP Researcher Spotlight: September 2023
The Monthly Researcher Spotlight is our section highlighting the exciting work of the UK Biobank ...

UK Biobank RAP Researcher Spotlight: March 2023
The Monthly Researcher Spotlight is our section highlighting the exciting work of the UK Biobank ...

UK Biobank RAP Researcher Spotlight: August 2023
The Monthly Researcher Spotlight is our section highlighting the exciting work of the UK Biobank ...

UK Biobank RAP Researcher Spotlight: July 2023
The Monthly Researcher Spotlight is our section highlighting the exciting work of the UK Biobank ...

UK Biobank RAP Researcher Spotlight: June 2023
The Monthly Researcher Spotlight is our new section highlighting the exciting work of the UK ...
Infrastructure Challenges: Machine Learning in the Cloud as a Future Oriented Solution Using UK Biobank
I will be contributing to a panel and an emerging session at this year’s Organization for Human ...

UK Biobank RAP Researcher Spotlight: April 2023
The Monthly Researcher Spotlight is our new section highlighting the exciting work of the UK ...

UK Biobank RAP Researcher Spotlight: February 2023
The Monthly Researcher Spotlight is our new section highlighting the exciting work of the UK ...

UK Biobank RAP Researcher Spotlight: January 2023
The Monthly Researcher Spotlight is our new section highlighting the exciting work of the UK ...

UK Biobank RAP Researcher Spotlight: May 2023
The Monthly Researcher Spotlight is our new section highlighting the exciting work of the UK ...
Announcing Enhanced Nextflow Support
At the recent Nextflow Summit in Barcelona, Product Manager and Principal Scientist, Dr. Yih-Chii ...

Celebrating a Birthday: The UK Biobank Research Analysis Platform Marks the First Anniversary of Its Launch
Oh, how quickly they grow! A year has passed since the launch of the UK Biobank Research Analysis ...

More Rapid Responses to Rare Disease
Families facing rare diseases know a thing or two about patience. They often face diagnostic ...

How Regeneron Bypasses Bottlenecks to Iterate at the Scale and Speed of Science
At the Regeneron Genetics Center (RGC), scientists are uncovering important genomic variants ...

How DNAnexus Enables Human Longevity to Optimize Genomics Analysis and Interpretation for Healthy Individuals - Inside DNAnexus
When the first draft of the human genome was completed in the early 2000s, the possibility of using ...

New Research Analysis Platform Enables UK Biobank To Exponentially Increase Size and Scale of World’s Most Comprehensive Biomedical Database - Inside DNAnexus
As the scope and data demands of the UK Biobank biomedical database gets bigger, we’re helping UK ...

The Hybrid Hackathons of the Future - now with Librarians! - Inside DNAnexus
Authors

Better Together: Regeneron & HLI Share Their Experiences with DNAnexus & AWS - Inside DNAnexus
Polymerase and primers. VCF and TBI. DNAnexus and AWS. Some things are just better together.

Visit us at Bio-IT World 2021! - Inside DNAnexus
We’re excited to see you back in you person at Bio-IT World in Boston! We’ll be there, masked up, ...

Computational Immunology: Tailoring Tools to Tackle the T-Cell Triad - Inside DNAnexus
Whether you’re a seasoned computational scientist or an armchair immunologist, the growing focus on ...

How New Practices in Biomedical Research are Changing the Future of Healthcare - Inside DNAnexus
Five years ago, we were ushering in the ‘era of precision medicine,’ with researchers and ...

How Multi-Omics is Changing Biomedical Research - Inside DNAnexus
Genomics, transcriptomics, proteomics, metagenomics… biology now seems to revolve around the ...

An Introduction to Population Genomics Studies with DNAnexus Apollo - Inside DNAnexus
While the amount and variety of genomics data available to scientists is greater now than ever ...
MIODx: A Breakthrough Leader in the precisionFDA COVID-19 Precision Immunology App-a-thon - Inside DNAnexus
The results are in for precisionFDA’s COVID-19 Precision Immunology App-a-thon, and we are thrilled ...
Responding to the Evolving World of COVID-19 Through Global Collaboration - Inside DNAnexus
Will the next wave of COVID-19 surveillance come via wastewater? What challenges lay ahead as we ...
DNAnexus Platform and Product Updates: 2020 in Review - Inside DNAnexus
Everyone is happy to see 2020 receding in the rearview mirror. But while last year was difficult in ...
Looking Beyond Causative Variants to Help Rare Disease Researchers Identify Elusive Treatment Options - Inside DNAnexus
Ever heard of schwannomatosis? How about neurofibromatosis? They are rare diseases that are not as ...
New UK Biobank Exome Pipeline: A 1-Button Read-Mapping Protocol for Public Sequencing Datasets - Inside DNAnexus
Public genomic resources such as UK Biobank are invaluable to researchers around the world. But the ...
2020 Vision: What Have We Learned? - Inside DNAnexus
The year 2020 certainly didn’t go as planned. But it was educational. We all had a crash course in ...
Deciphering the TCR Repertoire - Inside DNAnexus
Between the 4th and 19th centuries AD, knowledge of how to read and write Egyptian hieroglyphs was ...
DipAsm: A Method for Generating More Accurate Phased Assemblies - Inside DNAnexus
Researchers from academia and industry, including Li Lab (Dana-Farber Cancer Institute), Church Lab ...
NEW: Monitor your Diagnostics Pipeline with the Case Management Portal - Inside DNAnexus
If you’re running NGS data analysis pipelines for diagnostic purposes, then you know how important ...
MDA and DNAnexus Partner to Improve Neuromuscular Patient Care and Accelerate Drug Discovery - Inside DNAnexus
Outcomes for many with neuromuscular disease have improved dramatically in recent years, with the ...
From Pharmacogenomics to new Benchmarking Frameworks, DNAnexus Delivers at ASHG20 - Inside DNAnexus
Although we’ll miss the chance to spend some time in San Diego, we’re excited for the opportunity ...
How to Get Reliable Variant Calling in Repeat-Rich Regions - Inside DNAnexus
Current sequencing technology and computational algorithms support the construction of phased ...
Improving Therapeutic Development at Biogen with UK Biobank Data, Databricks, and DNAnexus - Inside DNAnexus
Genomic datasets from current day sequencing projects involve thousands of samples that routinely ...
Diving Deep into the Data Ocean - Inside DNAnexus
At City of Hope, POSEIDON was developed to manage its vast data ocean — an informatics platform fed ...
UK Biobank Democratizes Data Access with its own Cloud-Based Data Analysis Platform - Inside DNAnexus
Among the many lessons learned from the COVID-19 pandemic is that the world remains dangerously ...
How Myriad Genetics is Powering AI & Machine Learning Advancements in Precision Medicine - Inside DNAnexus
For nearly 30 years, Myriad Genetics has been at the forefront of precision medicine, transforming ...
Bold Innovation for a Promising Future - Inside DNAnexus
Running RStudio Shiny Server and Apps on DNAnexus - Inside DNAnexus
Shiny is an RStudio package for rapidly creating web apps in the R language. If your R script has ...
Embracing Open Standards to Build Portable, Reproducible Pipelines - Inside DNAnexus
It was Heraclitus who said “The only constant is change.” But it is the savvy bioinformaticians at ...
Three Ways to Leverage Translational Research for the Understanding of Complex Disease - Inside DNAnexus
Not so long ago, medical records were scribed on paper. A doctor’s office visit or hospital ...
2022 Forecast: Responding to the Rising Tide of Diagnostic Tests - Inside DNAnexus
Next-generation sequencing (NGS) has reached a turning point in diagnosing and treating rare and ...
Partnering with NVIDIA Clara Parabricks to Accelerate NGS Data Processing - Inside DNAnexus
Looking for ways to accelerate data processing and genomic analysis? By using graphics processing ...
Being Productive during COVID-19 Quarantine - Inside DNAnexus
A new Linus Group study finds that many life science practitioners have had their work disrupted, ...
A New Way to Play in May: Brush Up On Bioinformatics From Home - Inside DNAnexus
You told us what you wanted to learn, and we listened. Thanks to everyone who participated in our ...
Self-Servicing Archiving Now Available for DNAnexus on Azure - Inside DNAnexus
Whether you’re running DNAnexus on Amazon Web Services (AWS) or on Microsoft Azure, you can take ...
Truth V2 Challenge on precisionFDA: Calling Variants in Difficult-to-Map Regions using short, long, and linked read sequencing - Inside DNAnexus
Four years ago, precisionFDA, in collaboration with The Genome in a Bottle (GIAB) Consortium led by ...
Will COVID-19 Power a New Era of Precision Medicine? - Inside DNAnexus
As the cases of COVID-19 mount worldwide and scientists struggle to understand the new pathogenic ...
DNAnexus is going “virtual”! — Bio IT World 2020 - Inside DNAnexus
LIVE NOW! Visit the Wicked Smart digital experience: including webinars, expert demos, curated ...
Trending topics in bioinformatics/AI: a deep learning approach to antibiotic discovery - Inside DNAnexus
Working at DNAnexus has given me the opportunity to explore a bunch of AI/machine learning ...
Bioinformaticians for Good: DNAnexus Aids in Open Science Collaborations In Fight Against Coronavirus - Inside DNAnexus
Sequestered in bedrooms, kitchens, and makeshift office spaces around the world, a virtual army of ...
Our Commitment to our Customers and Partners During Coronavirus (COVID-19) - Inside DNAnexus
At DNAnexus, customer success is one of our core values and we take pride in delivering a high ...
BWA-MEM2 Review: Should You Upgrade? - Inside DNAnexus
Update June 11th, 2020: After our initial work for BWA-MEM2 evaluations, one of the developers, ...
Doubling Down on Next Generation Sequencing: How TwinStrand Biosciences and DNAnexus Work Together - Inside DNAnexus
In the majority of cancer patients, the first sign that something is wrong is finding an already ...
DNAnexus Platform Updates: A Year in Review - Inside DNAnexus
Now that we’re well into the new year, is everybody remembering to sign important documents with ...
DNAnexus Celebrates Rare Disease Day - Inside DNAnexus
The moment a baby is born is often described as the happiest day of a parent’s life. But imagine ...

Hackathon-ing into DNAnexus - Inside DNAnexus
Building a Precision Medicine Hub - Inside DNAnexus
Our understanding of human disease is progressing and so is the arc of precision medicine. ...
What Does the Sunsetting of Python 2.7 Mean for You? - Inside DNAnexus
As stated on python.org, the Python core development team sunset Python 2.x on January 1, 2020 and ...
Refining GWAS Results Using Machine Learning - Inside DNAnexus
A Tale of Two Cities: Breaking Down Data Barriers to Deliver Precision Medicine - Inside DNAnexus
Advances in genome sequencing technologies and the growing availability of clinical and other ...
Designing Bioinformatics Pipelines for Fast Iteration - Inside DNAnexus
When genetic tests are ordered, there’s probably little thought as to all of the bioinformatics ...
Providing Bioinformatics Solutions to Address Challenges with Structural Variants - Inside DNAnexus
Contributors: Arkarachai Fungtammasan, Jason Chin, Gigon Bae, Fernanda Foertter, Fritz Sedlazeck, ...
Addressing the Complex Storage and Archival Needs of DNA Sequencing Data - Inside DNAnexus
Computational biologist and large-scale computational DNA expert, Eric Schatz, estimates that by ...
Scaling to Meet the Demands of Rare Disease Genetics - Inside DNAnexus
Approximately 350 million people worldwide face the life-changing impacts of a rare disease. The ...
Building a GenomeArk on the Cloud - Inside DNAnexus
On August 28, 2019, the Vertebrate Genome Project (VGP) announced the completion of 100 ...
ASHG 2019: Delivering on the Promise of Precision Medicine - Inside DNAnexus
We have our bags packed and ready for Houston! The annual American Society of Human Genetics (ASHG) ...
DNAnexus Scoops European Innovations Award at SCOPE - Inside DNAnexus
We’ve always been proud of our innovative technology solutions, and recently we had the opportunity ...
Why Medical Centers Should Prioritize HITRUST in Cloud Providers - Inside DNAnexus
Considering an IT Investment For Your Clinical Diagnostics Lab? Help is Here - Inside DNAnexus
Next-generation sequencing data analysis management systems are the backbone of clinical ...
Examining Variation from Wet-Lab Protocol Choices in Microbiome Data through the Mosaic Standards Challenge - Inside DNAnexus
The Need for the Mosaic Standards Challenge Study of the human gut microbiome – the collection of ...
DNAnexus Detectives: Using Amazon Web Services to Help Solve a Medical Mystery - Inside DNAnexus
Our mission, and we chose to accept it, was to join more than 100 researchers and engineers to look ...
DNAnexus Navigation and UI Changes - Inside DNAnexus
To keep the DNAnexus platform easy enough for anyone to use and powerful enough for expert users, ...
By George! DNAnexus CTO George Asimenos Recognized as Top Voice in Precision Medicine - Inside DNAnexus
With his background in comparative metagenomics, silicon compilers, and elliptic curve ...
Another Feather in the Cap for PrecisionFDA Platform: A 2019 FedHealthIT Innovation Award - Inside DNAnexus
In the four years since its launch, the precisionFDA platform has helped foster collaboration, ...
Teaming Up To Improve Outcomes for Multiple Sclerosis Patients - Inside DNAnexus
Featured Webinar: A Novel Approach to Applying Genomic Patient Data to Improve Multiple Sclerosis ...
From Manhattan Plot to BigTop: DNAnexus Makes Data Visualization a (Virtual) Reality - Inside DNAnexus
Helping Scientists Discover the Hidden Jewels Within UK Biobank Data - Inside DNAnexus
Bio-IT World 2019: Explore Millions of Variants in the UK Biobank Data - Inside DNAnexus
We are heading back to our second home in Boston to attend the annual Bio-IT World conference! Our ...
Meet Us in Atlanta for AACR - Inside DNAnexus
We will be joining thousands of oncology researchers and clinicians this week at the American ...
Deep Dive into DeepVariant - Inside DNAnexus
Abstract In this blog, we analyze the non-deep learning components of DeepVariant in detail. We ...
Discussing the State of Biomedical Data & Looking Towards the Future at PMWC - Inside DNAnexus
DNAnexus once again attended the Precision Medicine World Conference (PMWC) in January in Santa ...
See our Talk at HiMSS 2019 - Integrating Clinico-Genomic Data for Precision Health - Inside DNAnexus
We’ve landed in Orlando and are ready to enjoy our time at the annual HiMSS Global Conference! In ...
HiMSS 2019 - Enabling Rapid Advancement of Precision Health & Drug Discovery at Scale - Inside DNAnexus
Highly Accurate SNP and Indel Calling on PacBio CCS with DeepVariant - Inside DNAnexus
PAG 2019: Pioneering Frontiers in Genome Assembly - Inside DNAnexus
DNAnexus is headed to San Diego! We’re excited to join over 3,000 leading genomic scientists in ...
2018 Highlights: Expanding Beyond Secondary Analysis and Scaling up Collaboration - Inside DNAnexus
We can’t believe it’s the end of the year already. 2018 has been extremely busy and productive year ...
Implementing an RNA-Seq pipeline on DNAnexus - Inside DNAnexus
Battling Aligners: Benchmarking Tools on Mosaic with Mock Metagenomes - Inside DNAnexus
DNAnexus at ASHG: Elevating Translational Informatics - Inside DNAnexus
Announcing the Results of Mosaic Clinical Strain Detection Challenge - Inside DNAnexus
Gabriel Al-Ghalith1, Sam Westreich2, Michalis Hadjithomas2
Parliament2: Fast Structural Variant Calling Using Optimized Combinations of Callers - Inside DNAnexus
Investigating Differences Between Simulated and Real FASTQ DNA-Seq Data - Inside DNAnexus
SMRT APAC Genome Assembly Grant & Upcoming Webinar - Inside DNAnexus
Submit your unique plant or animal genome proposal for a chance to win free de novo assembly ...
Breaking Down Crumble: A New Method to Significantly Reduce NGS Data Footprint while Preserving Results - Inside DNAnexus
New features for managing workflows and releasing them to your global network of collaborators - Inside DNAnexus
Comparison of BGISEQ 500 to Illumina NovaSeq Data - Inside DNAnexus
Announcing the Winners of Mosaic Microbiome Community Challenge: Strains #1 - Inside DNAnexus
The application of next-generation sequencing in the study of microbial communities has fueled the ...
PrecisionFDA Receives FDA Commissioner’s Award for Outstanding Achievement - Inside DNAnexus
Today, the precisionFDA Next Generation Sequencing (NGS) Team received the FDA Commissioner’s ...
SMRT Leiden Assembly Grant - Inside DNAnexus
Submit your unique plant or animal genome proposal for a chance to win free de novo assembly ...
Training and Applying Genomic Deep Learning Models - Inside DNAnexus
The application of Deep Learning methods has created dramatically stronger solutions in many ...
DNAnexus expands with Microsoft Azure Cloud Platform - Inside DNAnexus
At DNAnexus we focus on enabling scientific research and discovery by removing barriers such as ...
Bio-IT World 2018: Transforming Biological Insights to Accelerate Translational Research - Inside DNAnexus
Shaping the Future of Precision Medicine for Newborns - Inside DNAnexus
A Recap of Frontiers in Pediatric Genomic Medicine Conference
One Genome Browser to Rule Them All? - Inside DNAnexus
Amplifying Google's DeepVariant - Inside DNAnexus
We’re coming to the Windy City for AACR - Inside DNAnexus
We are excited to be attending our very first American Association for Cancer Research 2018 Annual ...
The Case for Rapid Genome Sequencing - Saving Critically Ill Newborns - Inside DNAnexus
GDPR Compliance - What you need to know - Inside DNAnexus
Regeneron Genetics Center Identifies Promising Target for Development of New Therapies for Chronic Liver Disease - Inside DNAnexus
How to Train Your DRAGEN - Evaluating and Improving Edico Genome's Rapid WGS Tools - Inside DNAnexus
Authors:
Integrating Multiple Data Sources to Power Discovery and Analysis - Inside DNAnexus
Precision Medicine World Conference (PMWC) took place in January in Mountain View, California, and ...
Advancing Microbiome Research through Community Engagement - Inside DNAnexus
The application of next-generation sequencing has transformed the study of microbial communities by ...
Benchmark and Advance Computational Methods for Targeted Strain Detection - Inside DNAnexus
The second Mosaic Community Challenge: Strains #2 is now live!
Tri-Con 2018: Leveraging the Cloud to Power Multi-Omics Research - Inside DNAnexus
The Molecular Medicine Tri-Conference kicks off next week in downtown San Francisco. Bringing ...
Countdown to AGBT 2018 - Inside DNAnexus
We can’t wait for the annual Advances in Genome Biology and Technology (AGBT) meeting, taking place ...
Analysis Commons: A Collaborative Approach to Multi-Omics Discovery - Inside DNAnexus
Advances in DNA sequencing have created large databases of whole-genome sequence (WGS) and ...
PMWC 2018: Leveraging Multi-Omic Datasets in Discovery & Clinical Trials - Inside DNAnexus
The Precision Medicine World Conference kicks off next week at the Computer History Museum in ...
Evaluating the Performance of NGS Pipelines on Noisy WGS Data - Inside DNAnexus
Dot: An Interactive Dot Plot Viewer for Comparative Genomics - Inside DNAnexus
Author: Maria Nattestad, Scientific Visualization Lead
Security Update: Meltdown and Spectre Vulnerabilities - Inside DNAnexus
On January 3rd, a new class of security flaw was reported that impacts most processors including ...
Gatk4 on DNAnexus - Inside DNAnexus
Authors:
PAG 2018: A Focus on Genome Assembly - Inside DNAnexus
The DNAnexus team is headed to San Diego, January 13-17, for the largest gathering of Ag-Genomics ...
CIO Webinar Series: Genomic Data Privacy in the Cloud - Inside DNAnexus
Join our two-part webinar series focusing on infrastructure requirements to scale geno-pheno ...
Evaluating DeepVariant: A New Deep Learning Variant Caller from the Google Brain Team - Inside DNAnexus
Launching Mosaic Community Challenge: Strains #1 - Inside DNAnexus
DNAnexus is excited to announce the launch of Strains #1, the first in a series of Mosaic Community ...
Mosaic: A DNAnexus-Powered Platform to Enable the Advancement of Microbiome Research - Inside DNAnexus
Please join us for the Mosaic app building webinar on Thursday, November 30, 2017 at 10am PST.
Comparison of Somatic Variant Calling Pipelines On DNAnexus - Inside DNAnexus
The detection of somatic mutations in sequenced cancer samples has become increasingly standard in ...
Introducing htsget, a new GA4GH protocol for genomic data delivery - Inside DNAnexus
DNAnexus is here in Orlando for the fifth plenary meeting of the Global Alliance for Genomics and ...
DNAnexus at ASHG: Accelerating Your Path from Genomic Data to Insight - Inside DNAnexus
We are looking forward to attending the annual American Society of Human Genetics (ASHG) meeting ...
Upgrading to TLS 1.2 - Inside DNAnexus
As a part of our continued efforts to maintain the highest security standards on the DNAnexus ...
Update to Ubuntu 14.04 - Inside DNAnexus
Ubuntu 12.04 has reached end-of-life, and the DNAnexus team is taking steps to ensure that customer ...
The New UI: Sleeker. Modern. Intuitive. - Inside DNAnexus
We are pleased to announce a design refresh of the DNAnexus web user interface! The new, modern ...
DNAnexus: Powering AZ's 2-Million Genome Translational Vision - Inside DNAnexus
The volume of biomedical data available for analysis is increasing at an exponential rate, yet ...
At Bio-IT World: Promoting Technological Innovation to Advance Precision Medicine - Inside DNAnexus
We are excited to join the 3,000+ researchers, clinicians, and pharmaceutical and IT professionals ...
Rady Children’s Quest to Finding That Needle in a Haystack - Inside DNAnexus
Rady Children’s Institute for Genomic Medicine (RCIGM), located in San Diego, has announced a ...
DNAnexus Celebrates National DNA Day - Inside DNAnexus
Known as National DNA Day, April 25th commemorates the discovery of the double helix structure by ...
Webinar: Trio Analysis with Sentieon Rapid DNAseq on DNAnexus - Inside DNAnexus
Please join us Thursday, May 18 at 11am PST (2pm EST) for an educational webinar on trio analysis ...
Upgrading to TLS 1.2 - Inside DNAnexus
At DNAnexus, security and compliance are paramount. In order to maintain the highest security ...
Case Study: Calling Somatic Variants with Sentieon on DNAnexus - Inside DNAnexus
Editor’s Note: This blog post was written by Don Freed, Bioinformatics Scientist at Sentieon. Email ...
ACMG: A Look at Applying Genomic Data to Clinical Reports - Inside DNAnexus
The annual American College of Medical Geneticists (ACMG) conference meets this week (March 21-25, ...
Updated DNAnexus Impact Assessment for Cloudbleed: No evidence of exploitation. - Inside DNAnexus
As described in our February 27, 2017 blog post regarding the Cloudflare information leak ...
Case Study: Trio Analysis with Sentieon Rapid DNAseq on DNAnexus - Inside DNAnexus
Editor’s Note: This blog post is written by Don Freed, Bioinformatics Scientist at Sentieon. Email ...
DNAnexus Not Impacted by Cloudflare Information Leak (“Cloudbleed”) - Inside DNAnexus
A serious bug within the code running on Cloudflare edge servers may have leaked sensitive data ...
Innovation Fueled by Collaboration and Regulatory Science - Inside DNAnexus
In mid-2015 the Food and Drug Administration’s (FDA) Office of Health Informatics awarded DNAnexus ...
New Org Admin Tools Available on DNAnexus - Inside DNAnexus
We are excited to announce the release of the Org Admin Interface, a new suite of tools to help ...
Sentieon on DNAnexus: License-Free Access Through April 7th - Inside DNAnexus
Test drive Sentieon’s pipelines on DNAnexus and see how you can achieve faster and more ...
Salute! Nexus Negronis On Us at AGBT! - Inside DNAnexus
The DNAnexus team is gearing up for the biggest genomics party of the year: AGBT. This annual ...
Bringing Together Genomics and Patient Data in the Cloud - Inside DNAnexus
Please join us Tuesday, February 7, at 10am PT (1pm ET) to hear leading genetics expert, Dr. ...
Collaborative Genomics: Highlights From 2016 - Inside DNAnexus
This has been a remarkable year for DNAnexus and the genomics industry at large. As 2016 comes to a ...
DNAnexus & TCGA: Reanalyzing the World's Largest Pan-Cancer Initiative Dataset - Inside DNAnexus
The Cancer Genome Atlas (TCGA), a joint effort between the National Cancer Institute (NCI) and the ...
Ten-Fold PacBio Sequel Call Set Proves Affordable & Effective in Identifying Structural Variants - Inside DNAnexus
Pacific Biosciences, a key partner of DNAnexus, has released the first public Sequel™ dataset of ...
Leading Genome Research Center Migrates to DNAnexus on Azure - Inside DNAnexus
Today we announced that the trusted DNAnexus genome informatics and data management platform is now ...
Join Us for a Lunchtime Discussion at ASHG - Inside DNAnexus
At DNAnexus we always look forward to attending the American Society of Human Genetics (ASHG) ...
Cloud Bioinformatics Made Easy: A Workshop with SCGPM - Inside DNAnexus
In April, the Stanford Center for Genomics and Personalized Medicine (SCGPM) adopted the DNAnexus ...
The U.S. Cancer Moonshot and a Culture of Collaboration - Inside DNAnexus
Yesterday, United States Vice President Joe Biden hosted the National Cancer Moonshot Summit. ...
Countdown to Beantown: DNAnexus at Festival of Genomics - Inside DNAnexus
Next week we’ll be at Festival of Genomics in Boston, to join the three-day celebration of all ...
Once in a Blue Moon Competition: precisionFDA Truth Challenge - Inside DNAnexus
The FDA, the Global Alliance for Genomics and Health (GA4GH) and National Institute for Standards ...
Launching the China Precision Medicine Cloud - Inside DNAnexus
Congratulations to WuXi AppTec, WuXi NextCODE and Huawei on launching their world-leading China ...
Frost & Sullivan Recognizes DNAnexus as the Enabling Technology Leader in the Global Genomics Industry - Inside DNAnexus
At DNAnexus, we’re honored to be recognized by Frost & Sullivan as one of the most significant ...
Webinar Series: Enabling PacBio Long-Read Bioinformatics in the Cloud - Inside DNAnexus
We are excited to be hosting, in collaboration with our partner PacBio, an inaugural webinar series ...
At Bio-IT World: Technological Innovation Advancing Genomic Science and Medicine - Inside DNAnexus
Bio-IT World Conference & Expo comes early this year (April 4-7th), where more than 3,000 ...
Precision Medicine from Birth - Inside DNAnexus
How many lives could be saved or significantly improved if precision medicine began at birth? Tute ...
One Global Cloud Platform Unites Researchers to Shed Light on Autism - Inside DNAnexus
Autism. It’s a word that evokes an inherently emotional response in many. And many wish for a cure, ...
The Journey to Precision Medicine - Inside DNAnexus
A Recap of the 9th Annual Future of Genomic Medicine Conference
Just Published New England Journal of Medicine Paper From Geisinger and Regeneron Highlights Value Of Integrating Genetic and EHR Data on DNAnexus - Inside DNAnexus
Traditionally, clinical genetic studies have involved deliberate recruitment of patients with ...
Cloud-based Genomics at the White House - Inside DNAnexus
The Launch of precisionFDA Consistency Challenge
The Future of Precision Medicine - Inside DNAnexus
Collaboration, Integration, Participation
Supporting Innovative Open Access Cancer Genomics Pilot - Inside DNAnexus
In January 2015, President Obama unveiled the Precision Medicine Initiative, an audacious research ...
DNAnexus Made Ridiculously Simple - Inside DNAnexus
In medical school, perhaps the most indispensable texts were the “Ridiculously Simple” series – ...
DNAnexus Heads to Orlando for AGBT 2016 - Inside DNAnexus
Each February, droves of scientists descend upon Florida for the annual Advances in Genome Biology ...
DNAnexus in Real Life: Tackling Precision Medicine Through Real-Time, Multi-Institution Cancer Collaboration - Inside DNAnexus
C. Anthony Blau, M.D. Today, the National Comprehensive Cancer Network (NCCN) announced an ...
A Look Back at 2015 - Inside DNAnexus
Before we officially welcome in the New Year, we can’t help but sit back and reflect on some of ...
DNAnexus Presents at JP Morgan Healthcare Conference - Inside DNAnexus
Today kicks off the 34rd annual JP Morgan Healthcare Conference, which continues to be healthcare’s ...
precisionFDA: Why It Matters - Inside DNAnexus
I hadn’t intended to write about precisionFDA going live – this post by Dr. Taha Kass-Hout and ...
Supporting Freebayes, to Serve Our Customers and the Community - Inside DNAnexus
Freebayes is a variant calling tool for short-read sequencing by Erik Garrison, Gabor Marth, and ...
Cancer Genomes Dataset Now Hosted on Amazon Web Services - Inside DNAnexus
Today, Amazon Web Services (AWS) and the Ontario Institute for Cancer Research (OICR) made ...
Highlights from Festival of Genomics California 2016 - Inside DNAnexus
PrecisionFDA, Explore Your Genome Giveaway, & Treadmills
Festival of Genomics: Not Your Typical Genomics Conference - Inside DNAnexus
Whole Genome Sequencing Giveaway, FDA’s New Approach to Regulatory Science & Treadmill Challenge
Post-ASHG: The Future of Genomics & Informatics in the Next 5 Years - Inside DNAnexus
Reference Genome Improvement, cfDNA for Transplantation, & de novo Reconstruction of Familial ...
Precision Medicine: Seeking Impact, Needing Champions - Inside DNAnexus
This weekend, I had the opportunity to present a short talk, entitled “Precision Medicine 2.0: ...
DNAnexus at ASHG: Supporting Diverse Applications from cfDNA Technology in the Clinic to High-Resolution Physical Genome Mapping - Inside DNAnexus
American Society of Human Genetics (ASHG) will be taking place in less than 2 weeks. One of the ...
New 3000 Rice Genomes AWS Public Dataset – Easy Access on DNAnexus Platform - Inside DNAnexus
In June we announced that DNAnexus was powering the 3000 Rice Genomes Project (3K RGP). You can ...
CareDx & DNAnexus Collaborate on Cloud-Based Genomics Platform for cfDNA Testing in Organ Transplantation - Inside DNAnexus
We are pleased to announce that CareDx has selected the DNAnexus cloud genomics platform to support ...
precisionFDA: A Community Approach for Submitting & Evaluating Diagnostic Tests, Powered by DNAnexus - Inside DNAnexus
DNAnexus has been awarded a research and development contract by the FDA’s Office of Health ...
Using Genomics To Improve Patient Care: DNAnexus to Support Both Projects Selected by California Precision Medicine Initiative - Inside DNAnexus
As Chief Medical Officer at DNAnexus, I’ve come to see my remit in very simple terms: I represent ...
Natera’s Genetic Testing to Scale with DNAnexus - Inside DNAnexus
Earlier this year, we announced that Natera, a leader in non-invasive genetic testing, selected the ...
The Rising Tide of Genomic Data Points to the Cloud - Inside DNAnexus
No other market segment has felt the profound impact of the cloud more than the life sciences ...
Help Save the Black Rhino - Inside DNAnexus
Ntombi the Rhino An alliance of institutions and individuals has formed to sequence the genome of ...
ENCODE Prepares For The Next Genome Data Explosion - Inside DNAnexus
A Challenging And Worthwhile Objective Last week ENCODE hosted a three-day Research Applications ...
DNAnexus Honored in MIT Technology Review's 50 Smartest Companies of 2015 - Inside DNAnexus
The first half of 2015 has proven to be a strong year for innovative companies in the biomedicine ...
DNAnexus at the Festival of Genomics - Inside DNAnexus
Will the inaugural Festival of Genomics (June 22-24, Boston) be like the psychedelic Woodstock ...
Precision Medicine Improves Survival without Increasing Costs - Inside DNAnexus
Today at the American Association for Cancer Research (AACR) Precision Medicine Series conference ...
Big Data Rice Research Helps to Feed the World - Inside DNAnexus
Supported by grants from the Bill and Melinda Gates Foundation and the Chinese Ministry of Science ...
DNAnexus Expands its Global Network for Genomic Medicine to China - Inside DNAnexus
It’s official – DNAnexus is expanding its cloud platform to China. A $15 million strategic ...
The Word “Genetics” is 110 Years Old - Inside DNAnexus
William Bateson William Bateson, an advocate of Gregor Mendel’s ideas, first proposed the use of ...
DNAnexus at Bio-IT World: Population-Scale NGS Studies Enter the Cloud - Inside DNAnexus
We are excited to be heading to Boston next week to participate in the annual Bio-IT World ...
NIH Security Best Practices Update - Inside DNAnexus
DNAnexus has always taken a proactive approach to security and compliance. We’ve worked closely in ...
A Signal Moment in Precision Medicine - Inside DNAnexus
The “Future of Genomic Medicine VIII” conference took place last week in beautiful La Jolla, ...
Events of Special Interest at the 16th Annual AGBT Meeting - Inside DNAnexus
It’s that time of year again when many genome scientists make their annual trip to one of the most ...
Obama’s Precision Medicine Initiative: DNAnexus is There - Inside DNAnexus
Last week, President Obama held a meeting unveiling details about the Precision Medicine ...
At PMWC 2015 – Keeping pace with NGS test volume & delivery - Inside DNAnexus
We are excited to be participating in the upcoming Silicon Valley Personalized Medicine World ...
Xconomy Forum: Healthcare’s Punctuated Equilibrium - Inside DNAnexus
Last week we had the pleasure of speaking at the Xconomy Forum: Innovation at Biotech’s Epicenter. ...
AWS Re:Invent – How Cloud Computing is Redefining Research - Inside DNAnexus
We were honored to receive an invitation to speak, together with Amazon Web Services (AWS) ...
A Sun Kissed Recap of ASHG 2014 - Inside DNAnexus
Last week, we had the pleasure of attending ASHG in sunny San Diego. This year’s meeting brought ...

100% Cloud-based Genome Center Integrating Large Healthcare Data Flows - Inside DNAnexus
photo: The Cancer Genome Atlas In a previous post, our new CMO, David Shaywitz, talked about his ...
Genomic & Phenotypic Data Collide at ASHG - Inside DNAnexus
It’s that time of year again! If you are headed to sunny San Diego for ASHG please be sure to stop ...
Harvesting Insights from the 2014 Plant Genomics Congress - Inside DNAnexus
Post Medicine X: Reflecting on a More Open Approach to Genomic Medicine - Inside DNAnexus
Last week, our Director of Science & Clinical Solutions, George Asimenos, Ph.D., gave a talk at ...
On A Day When Apple Sidesteps Healthcare Technology, Mary-Claire King Shows How To Confront It - Inside DNAnexus
The most interesting healthcare news of this week was manifestly not Apple AAPL +3.06%’s new watch; ...
At Stanford Medicine X: How Will Emerging Technologies Advance Healthcare? - Inside DNAnexus
We are very excited about the upcoming Stanford Medicine X conference that is taking place right ...
Towards Fulfilling The Promise Of Genomic Medicine - Inside DNAnexus
I was in eleventh grade when I first discovered The Eighth Day of Creation, Horace Freeland ...
The First Publicly Available “$1000 Genome” Test Dataset! - Inside DNAnexus
At DNAnexus we’re always looking for ways to collaborate on projects that are outside the norm, and ...
At ISMB, Talks Focused on Biological Discovery Through Computation - Inside DNAnexus
Recently, over 1,300 scientists converged in Boston to network, learn, and collaborate. They were ...
Techonomy Bio: A Provocative Look at Biotech Innovation - Inside DNAnexus
This week we attended the first-ever Techonomy Bio conference, a half-day series of discussions on ...
Discussing Clinical Genomics at the Recent ICCG Meeting - Inside DNAnexus
We had the pleasure of recently attending the International Collaboration for Clinical Genomics ...
What Will it Take to Make Big Data in Biomedicine a Success? - Inside DNAnexus
Last week, we attended the Big Data in Biomedicine conference which was held in our backyard at ...
Congratulations, Team Baylor! - Inside DNAnexus
This week our friends and collaborators at Baylor College of Medicine’s Human Genome Sequencing ...
DNAnexus Introduces Faster Cloud Options - Inside DNAnexus
Spring has arrived at DNAnexus, ushering in important updates! Starting May 1, 2014, we are excited ...
Back to Beantown: Kicking off Bio-IT World - Inside DNAnexus
This week the DNAnexus team is heading to Boston for the annual Bio-IT World Conference & Expo, ...
DNA Day: Celebrating the Decades-long Unraveling of DNA - Inside DNAnexus
Security Advisory: Response to Heartbleed Vulnerability - Inside DNAnexus
On April 7, 2014, a serious vulnerability known as Heartbleed (CVE-2014-0160) was disclosed in the ...
AGBT Posters Featuring DNAnexus - Inside DNAnexus
DNAnexus was featured in a couple of posters at AGBT this year. DNAnexus scientist, Andrew Carroll, ...
Feb 28: Celebrating Rare Disease Day - Inside DNAnexus
A person with Alport Syndrome might have to look far and wide to find somebody else who can truly ...
AGBT Conversation Centers on Data Analysis and Computational Challenges - Inside DNAnexus
Marco Island, Here We Come! - Inside DNAnexus
February marks the time scientists throughout the genomics community make the annual pilgrimage to ...
2014: The Year of the Cloud - Inside DNAnexus
Bring on the New Reference Genome! - Inside DNAnexus
Like many of our fellow genomics scientists, we are eager to see the much-anticipated new human ...
One Simple Solution for Ten Simple Rules - Inside DNAnexus
Like many in the systems biology space, we have been longtime fans of Philip Bourne’s Ten Simple ...
CHARGE-ing Ahead After ASHG - Inside DNAnexus
After last week’s ASHG frenzy, we could use a week off! But we’re so inspired by the positive ...
Run the Mercury Variant-Calling Pipeline on Your Own Data - Inside DNAnexus
Mercury, designed by the Human Genome Sequencing Center at Baylor College of Medicine (HGSC), is ...
See You at ASHG — Lunch Is on Us! - Inside DNAnexus
We are looking forward to catching up with you at ASHG. You can stop by booth #915 to check out the ...
At Beyond the Genome, Talks Focused on Single-Cell Genomics and Informatics - Inside DNAnexus
Beyond the Genome, a top-notch conference hosted by Genome Biology and Genome Medicine, took place ...
The Hundred Year Study? Newborn Sequencing Grants Bring Opportunity for Long-Term Data Analysis - Inside DNAnexus
The announcement last month that the National Human Genome Research Institute and National ...
News: DNAnexus Offers Service for Clinical Testing Labs - Inside DNAnexus
This week we’re pleased to announce the launch of our specialized platform-as-a-service (PaaS) for ...
Keep Your HIPAA-Protected Data Safer with Cloud Computing - Inside DNAnexus
If you’ve been considering the implications of cloud computing when it comes to HIPAA compliance, a ...
New Single-Cell Genomic Studies Demonstrate Utility of SPAdes Assembler - Inside DNAnexus
This summer we saw some new publications underscoring the need for a high-quality assembler for ...
Cloud Computing Insight with Omar Serang - Inside DNAnexus
ISMB Recap: Looking for Knowledge and Imagination - Inside DNAnexus
Earlier this week we were lucky enough to attend the annual Intelligent Systems for Molecular ...
Developer Spotlight: A De Novo Assembler Named Ray - Inside DNAnexus
We recently launched the DNAnexus developer program, and to our delight one user was able to ...
Looking Forward to ISMB and Meeting Developers in Berlin - Inside DNAnexus
It’s July, and you know what that means — we’re getting ready for the annual Intelligent Systems ...
Looking Back at SFAF in Santa Fe - Inside DNAnexus
We recently attended the Sequencing, Finishing, and Analysis in the Future (SFAF) meeting, hosted ...
Publication Watch: In Early 2013, Nice Flow of New Papers from DNAnexus Users - Inside DNAnexus
It’s been awhile since we checked in on publications using DNAnexus, so we headed over to PubMed to ...
On DNA Day, We're Thinking About (What Else?) Data - Inside DNAnexus
Today is DNA Day! This year it’s an especially big deal as we’re honoring the 60th anniversary of ...
At Bio-IT World, Genome Centers Dished on Big Data - Inside DNAnexus
At the Bio-IT World Conference & Expo last week in Boston, more than 2,500 attendees descended ...
Join Us at Bio-IT World for a Personalized Demo and Chance to Win an iPad Mini! - Inside DNAnexus
Next week kicks off the annual Bio-IT World Conference & Expo, one of the best conferences ...
Meet the new DNAnexus and its Instant Collaboration Environment - Inside DNAnexus
The life sciences field has a long and respected tradition of collaboration among researchers. ...
Recovering from AGBT: Exhausted but Encouraged ! - Inside DNAnexus
It’s hard to believe the whirlwind of the annual Advances in Genome Biology & Technology ...
Meet the new DNAnexus and Its Extensible Genomics Toolbox - Inside DNAnexus
This week we continue our look at unique facets of the new DNAnexus with a focus on the “Extensible ...
What to Pack for AGBT 2013: Sunblock, Flip-flops, and Data ! - Inside DNAnexus
Sequencing experts around the world are honing their slide decks, performing their last data ...
Meet the new DNAnexus and its Configurable Cloud Infrastructure - Inside DNAnexus
It’s been a busy first week since we launched the beta of the new DNAnexus, our cloud-based DNA ...
Hello World! Come Take the new DNAnexus for a Test Drive - Inside DNAnexus
Today marks the official beta launch of our new platform, and we can’t wait for you to try it out!
JP Morgan in Review: Expect Rapid Evolution in Sequence Analysis and Big Data Needs - Inside DNAnexus
Last week’s JP Morgan Healthcare Conference was the usual biotech extravaganza, filling downtown ...
Dispelling the Myths of the Cloud - Inside DNAnexus
What comes to mind when you hear the word “cloud”? Does the Amazon cloud immediately pop into your ...
Happy Holidays to All! - Inside DNAnexus
DNAnexus in the Literature: A Look at Recent Papers Using Our Platform - Inside DNAnexus
It’s great to find that several papers published recently have used DNAnexus in their research — ...
At NGS Meeting: Epigenetics & "Dark Matter" Were Major Themes - Inside DNAnexus
It was with great enthusiasm that I attended Oxford Global’s recent 4th Next Generation Sequencing ...
ASHG: With Great Science Comes Great Responsibility - Inside DNAnexus
The American Society of Human Genetics annual meeting was held at the Moscone Center in San ...
Join DNAnexus for a Lunchtime Workshop at ASHG - Inside DNAnexus
Whether you join us for lunch to explore the dark matter of ENCODE or visit our booth (#507) to ...
Collaborative Research Was the Big Winner at Bio-IT World Europe - Inside DNAnexus
At Beyond the Genome Conference, Lessons on Data Analysis and Clinical Studies - Inside DNAnexus
New ENCODE Paper Reveals Remarkable Chromatin Diversity at Regulatory Elements - Inside DNAnexus
Today marks a major milestone for the ENCODE consortium! More than 30 papers will be published ...
At Rhode Island NGx Conference, Informatics Is Clearly on the Rise - Inside DNAnexus
We are back from Cambridge Healthtech Institute’s NGx Next-Generation Sequencing Data Analysis ...
At ESHG, Clinical Utility Steals the Spotlight - Inside DNAnexus
The annual European Human Genetics Conference, better known as ESHG for its sponsor, the European ...
At Bio-IT World, All Eyes Were on the Cloud and Big Data! - Inside DNAnexus
As expected, the 10th annual Bio-IT World Conference & Expo was both exhausting and ...
Hope to See You at Bio-IT World! - Inside DNAnexus
Here at DNAnexus, we’ve been gearing up for the 10th annual Bio-IT World Conference & Expo, ...
Scientific Collaborators in New York and Jerusalem Uncover New Mutation Underlying Rare Sensory Disease - Inside DNAnexus
The study described below was published in the April 2012 edition of the Annals of Neurology, the ...
On the Scene at AMIA: Clinical Promise and Informatics Opportunities for Whole-Genome Sequencing - Inside DNAnexus
I recently got to attend the American Medical Informatics Association’s (AMIA) Joint Summits on ...
ABRF: A Quick Meeting Recap - Inside DNAnexus
Here at DNAnexus, we’re lucky to have a terrific team supporting our goals. In this blog post, we ...
SOT: Still Early Days for Next-Gen Sequencing in Molecular Toxicology - Inside DNAnexus
The Society of Toxicology’s 51st annual meeting was held this week right in our back yard. Since I ...
On Being Platform Agnostic - Inside DNAnexus
One inevitable outcome of the ever-expanding number of DNA sequencing platforms is the lock-step ...
AGBT in Review: Highlights and High Hopes for Data - Inside DNAnexus
Last week’s Advances in Genome Biology and Technology (AGBT) meeting was every bit the fast-paced ...
Relationships for Innovation - Inside DNAnexus
This week we announced new agreements with two premier healthcare institutions: Geisinger Health ...
Load Up on Caffeine ... AGBT Is Almost Here - Inside DNAnexus
View from the Marcos Island Marriott, the AGBT venue We’re gearing up for the Super Bowl of the ...
Sequence Data: The View from JP Morgan - Inside DNAnexus
Last week, Andrew Lee, Vice President of Strategic Operations, and I attended the JP Morgan ...
Preserving and Enhancing an Important Community Resource - Inside DNAnexus
Today, DNAnexus is pleased to announce the launch of our hosted SRA site! The DNAnexus SRA site is ...
Taking DNAnexus to the next level - Inside DNAnexus
Today we’re proud to announce the addition of two partners to DNAnexus: Google Ventures and TPG ...
Case Study Highlight: Differential Expression of Splicing Factor Genes - Inside DNAnexus
Dr. Miriam Bucheli, an instructor at Harvard University, is this month’s use case focus. She ...
Seeing The Trees In The Forest - Inside DNAnexus
One of the biggest challenges associated with the identification of genomic variation, is finding ...
Streaming an entire sequencing center across the Internet - Inside DNAnexus
How much next-gen sequencing data do the top genome centers in the world produce? It’s a staggering ...
Navigating the Exome with DNAnexus - Inside DNAnexus
With a growing number of targeted exome capture solutions being integrated into the next-generation ...
Next-gen sequencing and the cloud - revolutionary, or hype? - Inside DNAnexus
It’s been an exciting time for DNAnexus since launching our company at the recent Bio-IT World ...