

Update June 11th, 2020:
After our initial work for BWA-MEM2 evaluations, one of the developers, Vasimuddin Md (@wasim_galaxy), had contacted us about a number of improvements and shared with us the latest BWA-MEM2 binary on May 22nd, 2020. We have some updated information to share after testing out the suggested version and running parameters.
With the latest binary, we revisited our discrepancy analysis between BWA-MEM and BWA-MEM2 with the three samples (SRR10150660, SRR10286881, and SRR10286930) that had the largest differences in our original post. We found out that the mapping results of BWA-MEM2 are exactly the same with BWA-MEM using this latest version.
The identical result is discovered when aligning against both hg38 builds, hs38DH (GRCh38 primary contigs + decoy contigs + ALT contigs and HLA genes), as well as GCA_000001405.15_GRCh38_no_alt_analysis_set (GRCh38 primary contigs + decoy contigs, but no ALT contigs nor HLA genes). Additionally, while the per sample wall clock runtime is slightly longer with a range of 10%, BWA-MEM2 still gives the advantage of being around ~2X faster to its precedence, BWA-MEM, as we have found originally.
Additionally, three of our previously failed BWA-MEM2 analysis, which were reported on bwa-mem2 github page (35, 49, and 50), can now be finished successfully by utilizing a lower number of thread usage (with bwa-mem2 option -t), or by increasing the memory capacity.
BWA-MEM2 — the Intel accelerated version of BWA-MEM
The Burrow-Wheeler Aligner (BWA-MEM), which requires no introduction, is one of the most popular software tools in the Bioinformatics and Genomics industry. Being the first step short-reads undergo after generated by a sequencing instrument, BWA-MEM has been widely used as a common upstream tool. BWA-MEM generates alignments to a reference genome for a variety of germline and somatic genetic variant detections, such as single-nucleotide polymorphisms (SNPs), indels, structural variations, and copy number variations. With the widespread use of next-generation sequencing technologies, more and more sequencing reads are now being generated for translational research and clinical diagnostics. Scientists need to be able to process these reads more efficiently and cost effectively.
BWA-MEM2, which was recently announced at ipdps2019 (2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS)) and published[1] by Vasimuddin Md (@wasim_galaxy), Sanchit Misra (@sanchit_misra), Heng Li (@lh3lh3), and Srinivas Aluru from the Intel Corp., Dana-Farber Cancer Institute and Georgia Institute of Technology. The team optimized algorithms of BWA-MEM for three key kernels: (1) Search for super maxima exact matches (SMEM), (2) Suffix array lookup (SAL), and (3) banded Smith Waterman (BSW) algorithm. Part of the optimizations utilized the AVX-512 instruction, which is for the Intel® Xeon® Skylake processor and an Intel® Xeon® E5 v3 processor, while the other optimizations are generic and can provide performance gains on any of the modern processors.
In this blog post, we would like to share our experiences on performing BWA-MEM2 with the next-generation instance types (c5d) that use the Intel Xeon Scalable Processors (Cascade Lake) or Intel Xeon Platinum 8000 series (Skylake-SP), and utilize the AVX-512 instruction set on a cloud computing environment. We also are demonstrating the comparisons between running BWA-MEM and BWA-MEM2.
Mapping with BWA-MEM vs. BWA-MEM2
To represent reads from a range of different sequencing instruments, library prep, and sequencing labs, we downloaded paired-end FASTQ files from Sequence Read Archive (SRA) by randomly drawing ten samples from each of the Illumina HiSeq 2500, HiSeq 4000, HiSeq X Ten, and NovaSeq 6000. Five of the samples were from whole exome sequencing (WES) library prep and another five were from whole genome sequencing (WGS) library prep, according to their SRA records. All of the samples were submitted to SRA from January to November 2019.
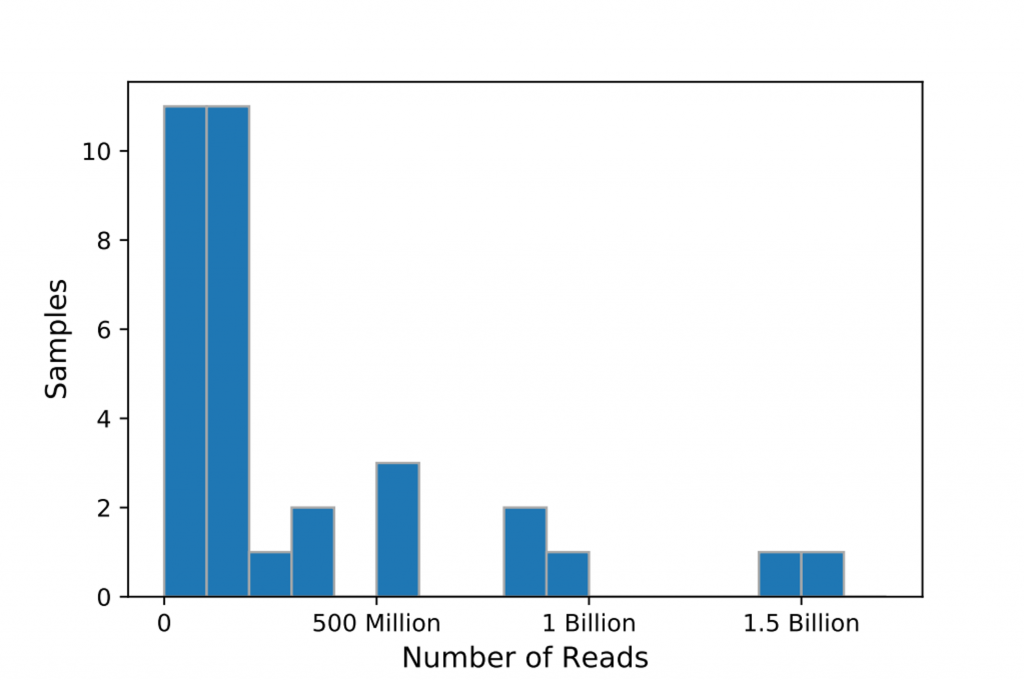
Five out of the 40 samples we drew didn’t get resolved into paired-end FASTQ files. For simplicity, we discarded the five to benchmark alignment without paired-end reads, which is the most common use case at DNAnexus. The remaining 35 paired-end samples consist of 307 million to 1.59 billion reads. All samples have read lengths distributed from 100bp to 151bp, except one sample (SRR10028120) has its shortest read down to 18bp. Figure 1 shows the distribution of number of reads across samples.
We mapped reads against hs38DH which consists of primary contigs + ALT contigs + decoy contigs and HLA genes from the GRCh38 assembly. For performing BWA-MEM, we used the BWA-MEM FASTQ Read Mapper app, which runs BWA-MEM v.0.7.15 while piping the mapped reads into biobambam2 version 2.0.87 for coordinate sorting and marking duplicates. For BWA-MEM2, we used the same pipeline configuration but swapped the BWA-MEM binary to BWA-MEM2 binary built from 1038fe3. Each sample was processed on a single AWS c5d.9xlarge instance utilizing full 36 vCPUs, and the I/O was managed on the DNAnexus Platform.
Commands we used for running BWA-MEM and BWA-MEM2:
# BWA-MEM: bwa mem -t 36 \ /path/to/hs38DH.fa \ /path/to/SAMPLE_R1.fastq.gz \ /path/to/SAMPLE_R2.fastq.gz \ -K 100000000 -Y \ -R '<SAMPLE Read Group INFO>' \ | bamsormadup SO=coordinate threads=36 level=6 inputformat=sam indexfilename=SAMPLE_NAME.bam.bai M=SAMPLE_NAME.duplication_metrics # BWA-MEM2: bwa-mem2 mem -t 36 \ /path/to/hs38DH.fa \ /path/to/SAMPLE_R1.fastq.gz \ /path/to/SAMPLE_R2.fastq.gz \ -K 100000000 -Y \ -R '<SAMPLE Read Group INFO>' \ | bamsormadup SO=coordinate threads=36 level=6 inputformat=sam indexfilename=SAMPLE_NAME.bam.bai M=SAMPLE_NAME.duplication_metrics
Findings
BWA-MEM2 is actively WIP (work in progress)
With the 35 samples we processed, three of them failed in BWA-MEM2, while they all were mapped smoothly with BWA-MEM. We have reported these events to the bwa-mem2 github page (35, 49, and 50). They were all addressed by Intel’s Parallel Computing Lab prior to this blog posting.
BWA-MEM2 is ~2X faster
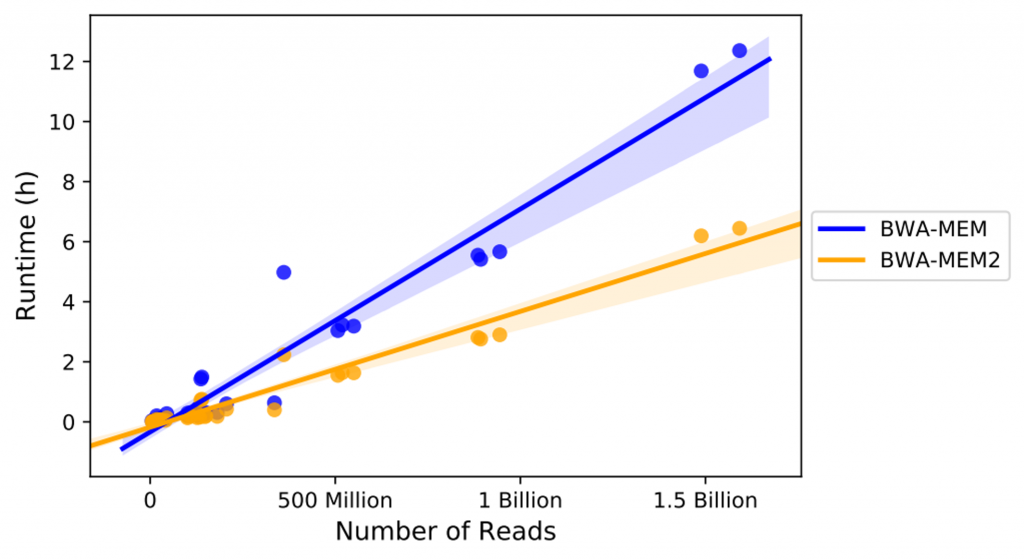
In Figure 2, we collected the walk-clock runtime of alignment for all 32 samples that mapped successfully using both BWA-MEM and BWA-MEM2 in multi-threading mode (36 threads). The runtime includes pipeline setup, mapping, sorting, markdup, as well as I/O between a c5d.9xlarge instance and an s3 bucket. Note that we deducted time spent for transferring the reference genome index onto the instance, because the significant larger size of the reference genome index file for BWA-MEM2 (30.27 GiB) comparing to 3.32GiB for BWA-MEM, which roughly took 8 mins and 48 seconds per sample run, respectively.
Not surprisingly, we found the runtime scales linearly with the number of reads for both BWA-MEM and BWA-MEM2. On average, BWA-MEM2 runs 2.07 times faster than BWA-MEM, while the standard deviation of the runtime ratios is 0.66. This trend is similar to what was found in BWA-MEM2’s performance report (link). With the ~2X acceleration, BWA-MEM2 not only significantly reduces the turnaround time from reads off the sequencing instrument to clinical diagnosis results, but also provides a more cost-effective solution for read alignment.
Discrepancies in BWA-MEM and BWA-MEM2
We compared the mapping results from the two tools to validate if BWA-MEM2 is reproducing the identical mappings as BWA-MEM and to evaluate if there are any discrepancies that could impact downstream genetics variants detections.
We used BamUtil to compare the mapped, sorted and duplicate marked BAMs from BWA-MEM and BWA-MEM2. We counted a read as being reported differently when a read was mapped to different positions, or having different tag values in MD:Z (string for mismatching positions), NM:i (edit distance to the reference), MQ:i (mapping quality of the mate/next segment), RG:Z (readgroup), XA:Z (alternative hits), or XS:i (Suboptimal alignment score).
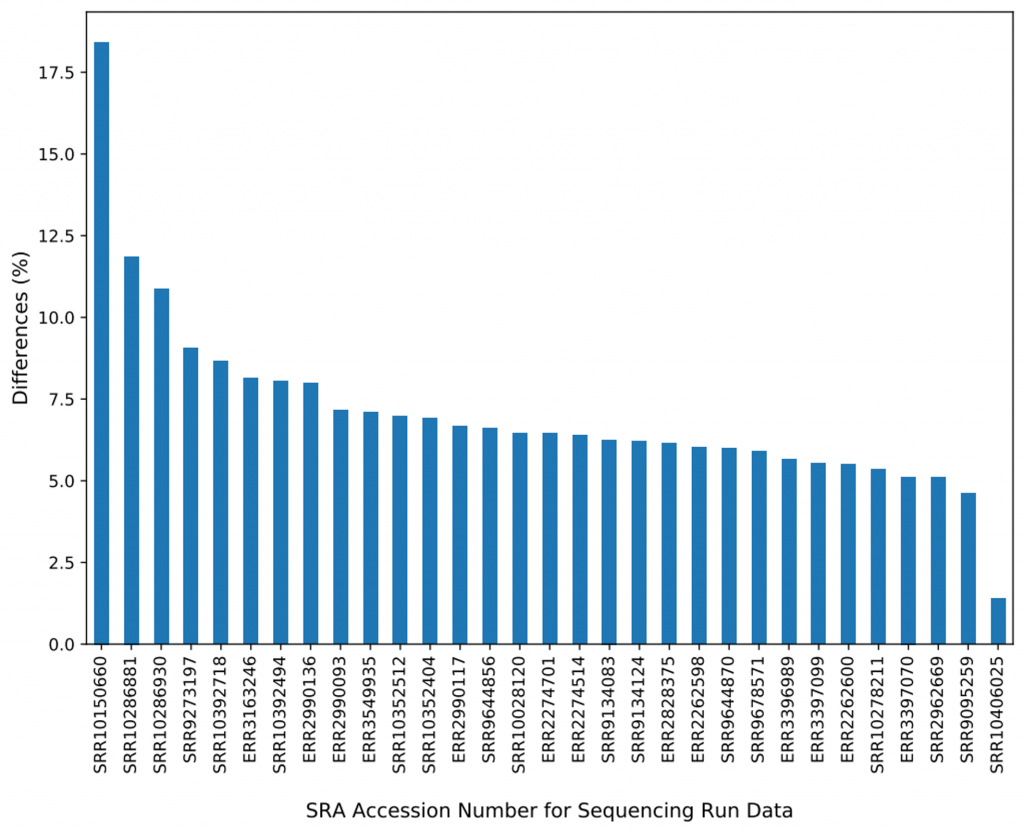
Two (SRR10270814 and SRR10270774) out of the 32 samples were found to have large amounts (89% and 93%) of discrepancies between the BAMs from BWA-MEM and BWA-MEM2. We realized that the two samples were sequence reads of vancomycin-resistant Enterococcus faecium (VREfm) isolated from stool and blood cultures of patients undergoing chemotherapy or hematopoietic stem cell transplantation, despite the fact we limited our sampling search for only Homo sapiens organisms. We removed these two samples from further comparisons. For the remaining 30 samples, on average, 7.11% of the mapped reads are reported differently in BWA-MEM and BWA-MEM2. Figure 3 shows the percentage differences of reads reported per sample in BWA-MEM.
Where are the differences from? ALT contigs.
For all samples, around half of the differences are either exclusively classified by BWA-MEM or BWA-MEM2. The other half of differences were classified by BWA-MEM reporting all alternative mapping locations (while BWA-MEM2 does not report any of those locations), mate-mapping quality score, suboptimal alignment score, mapping string, CIGAR string, and others (Figure 4).
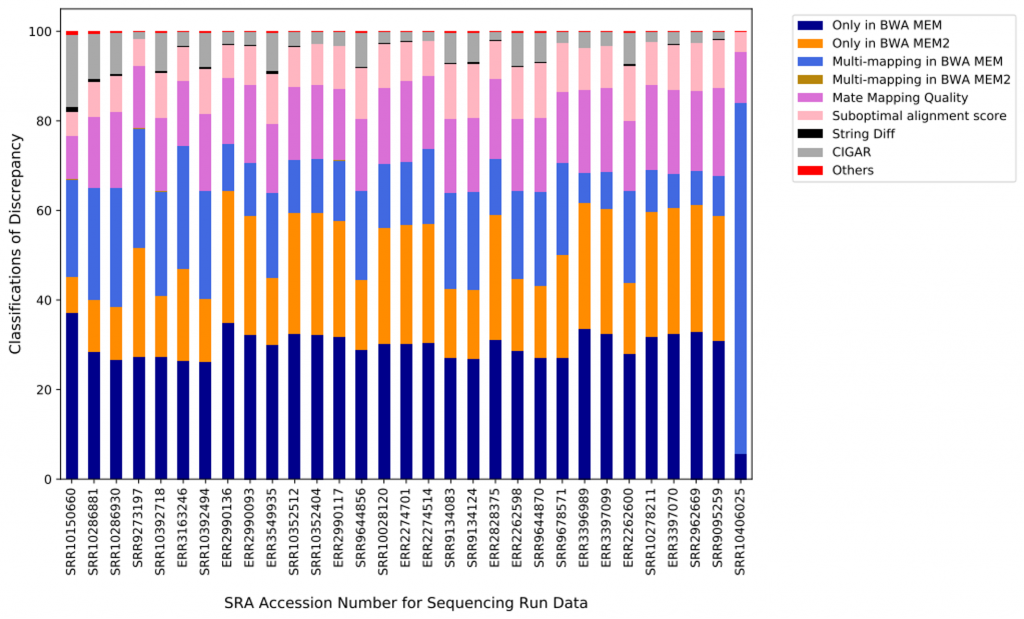
We were surprised about the high number of differences because concordance is crucial for all downstream variant calling analysis. We consulted this discovery with Heng Li, and followed up by repeating the same comparison for the three samples (SRR10150660, SRR10286881, and SRR10286930) that differ the most — this time we mapped the reads against GCA_000001405.15_GRCh38_no_alt_analysis_set, which consists GRCh38 primary contigs and human decoy region (hs38d1), but no ALT contigs nor HLA genes. The differences between these three samples mapped by BWA-MEM and BWA-MEM2 dropped from 11%–18% down to all being less than 0.001%, which probably has a negligible effect. We suspected that mapping to ALT contigs and HLA genes may trigger some corner cases which were not yet properly implemented in BWA-MEM2.
While variant calling for reads mapped to ALT contigs is not yet widely taken for downstream software tools, we look forward to the equivalent ALT contig and HLA-gene alignments in BWA-MEM2 to BWA-MEM.
Final thoughts & discussions
The command line interface of BWA-MEM2 is the same as BWA-MEM — we used the exact same syntax for specifying inputs (reads and the reference genome) and setting parameters. Additionally, the output SAM format is compatible with downstream software tools (i.e. samtools and bamsormadup). In addition, BWA-MEM2 seamlessly determines and runs the most efficient mode among AVX512, AVX2, SSE4.1, or Scalar, depending on what is the fastest mode possible with the instruction set architecture of the instance.
BWA-MEM2 requires a larger reference genome index file. Take hs38DH as an example, it takes up to 30.27GiB storage space in gzipped tarball (tar.gz) format. This is almost one order of magnitude bigger than the same genome index for BWA-MEM (3.32GiB). In some settings for the cloud computing environment, this can cause additional runtime overhead to set up the alignment environment, such as moving the genome index to the instance and extracting the tarball. We also ran into cases while a sample requires more than the memory limit of the c5d.9xlarge instance’s memory capacity (72GiB) and ran out of memory. During our discussion with the Intel team via Github, we learned that we could lower the threads in use with the cost of slower mapping. We look forward to possible future BWA-MEM2 updates to reduce the memory footprints for certain scenarios.
Should you upgrade? BWA-MEM2 shows significant advancement with its 2X speedup, compatible I/O interface to BWA-MEM, seamless support for different chip instructions, and equivalent mapping results as BWA-MEM. While it is provided as “not recommended for production uses at the moment” by the Intel team, we expect to see these short-read alignment upgrades to BWA-MEM2 in the foreseeable future.
Acknowledgements
We’d like to thank Heng Li for his valuable feedback of the BWA-MEM and BWA-MEM2 comparison work and insights about reference genome builds. In addition, we’d like to thank Brett Hannigan (@gildor) for the initial design and discussion of this work.
References
[1] M. Vasimuddin, S. Misra, H. Li and S. Aluru, “Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems,” 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS), Rio de Janeiro, Brazil, 2019, pp. 314-324.


.png)
.png)
.png)