

In this blog we quantify Google Brain’s recent improvements to DeepVariant – detailing significant improvement in both exomes and PCR genomes. We reflect on how this improvement was achieved and what it suggests for deep learning within bioinformatics.
Introduction
The Google Brain Team released DeepVariant as an open-source GitHub repository in December 2017. Our initial evaluation of DeepVariant and our Readshift evaluation (blog) method (code) identified that while DeepVariant had the highest accuracy of all methods on the majority of samples, there were a few outliers with much higher Indel error rates.
Subsequently, we realized that these difficult samples had a common feature – they were all prepared with PCR. Also, the initial release of DeepVariant had not been trained on exome data. Based on this, DNAnexus provided exome and Garvan Institute PCR WGS samples to Google Brain to train improved DeepVariant models, which are now released. We also recognize Brad Chapman, who independently collaborated with Google Brain.
Improving Deep Learning Methods Compared to Improving Traditional Methods
Many factors make variant calling more complex in PCR+ samples and exomes. PCR amplification is biased by GC-content and other factors. Errors that accumulate in the PCR process are difficult to distinguish from true variants. Exomes have an additional complexity of uneven capture efficiency and coverage.
Knowledge of these facts is incorporated differently in human-programmed methods, traditional machine learning, and deep learning methods. Human-written methods require a programmer to carefully develop heuristics which capture the relevant properties without too much rigidity. Traditional machine learning approaches require a scientist to identify informative features that capture the information, encode those from the raw data, and train and apply models.
Deep learning based methods like DeepVariant attempt to represent the underlying data in as raw a manner as possible, relying on the deep learning models to learn the features. Here, the scientist’s role becomes to identify the examples that embody the diversity of conditions the tool must solve, and to organize how to present as many and varied of these examples to the training machinery as possible. How Google Brain made these improvements may be as interesting as the improvements themselves.
Exome-Trained Models Significantly Improve Performance
Google Brain’s initial release was DeepVariant v0.4. The v0.5 version added the ability to apply either a WGS model or an exome model. The v0.6 version specifically added PCR+ training for the first time.
All evaluations use the hs37d5 reference and v3.3.2 Truth Sets from NIST Genome in a Bottle. Evaluation is performed with Hap.py in the same method used in PrecisionFDA. Google’s release notes indicate they never train on HG002 or chr20 of any sample.
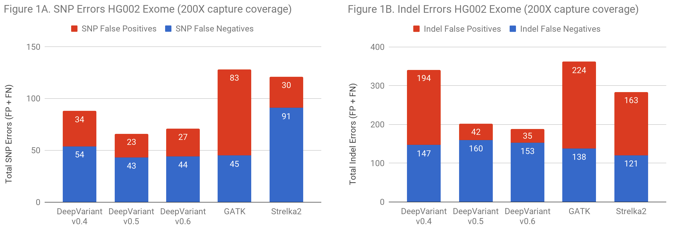
On exomes, DeepVariant improved from approximate parity to a 2-fold error reduction relative to GATK. There are two ways to put this improvement in accuracy into context. DeepVariant has an absolute decrease of 231 errors and 121 false positives. For a callset of 500,000 exomes – as the UK BioBank will soon make available – the fact that false calls tend to distribute randomly while true variants are shared in the population could mean a reduction of 60 million false variants positions in the full callset (for reference, the 60,000 exome ExAC set contained 7 million variant positions).
Another way to frame the value is: does the improved accuracy allow similar performance as traditional methods at a lower coverage?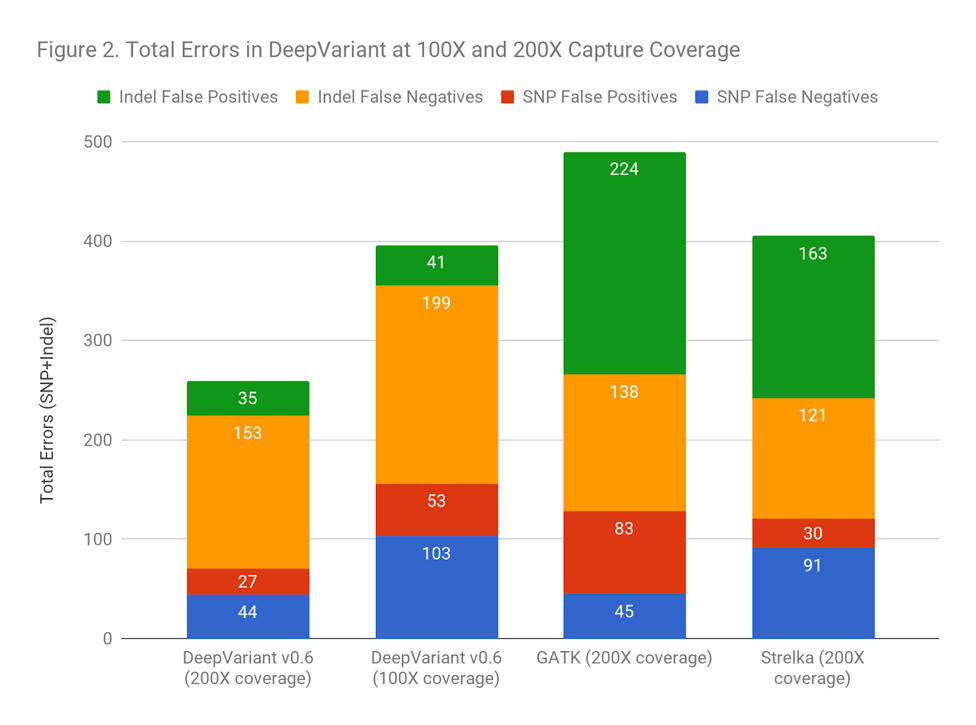 Figure 2 demonstrates that DeepVariant on the same exome downsampled randomly to 50% coverage is more accurate than other methods at full coverage. This could allow a re-evaluation of required sequencing depth to either increase throughput, save cost, or refocus sequencing depth on complex but important regions.
Figure 2 demonstrates that DeepVariant on the same exome downsampled randomly to 50% coverage is more accurate than other methods at full coverage. This could allow a re-evaluation of required sequencing depth to either increase throughput, save cost, or refocus sequencing depth on complex but important regions.
Including PCR+ WGS Data Greatly Improves DeepVariant Performance
DeepVariant was not specifically trained on PCR-prepared WGS samples until the v0.6 release, when 5 PCR+ genomes were added to the training set. Even before this addition, DeepVariant was the most accurate SNP caller surveyed in our PCR+ data. However, as Figure 3A indicates, the inclusion of PCR training data improved SNP accuracy noticeably.
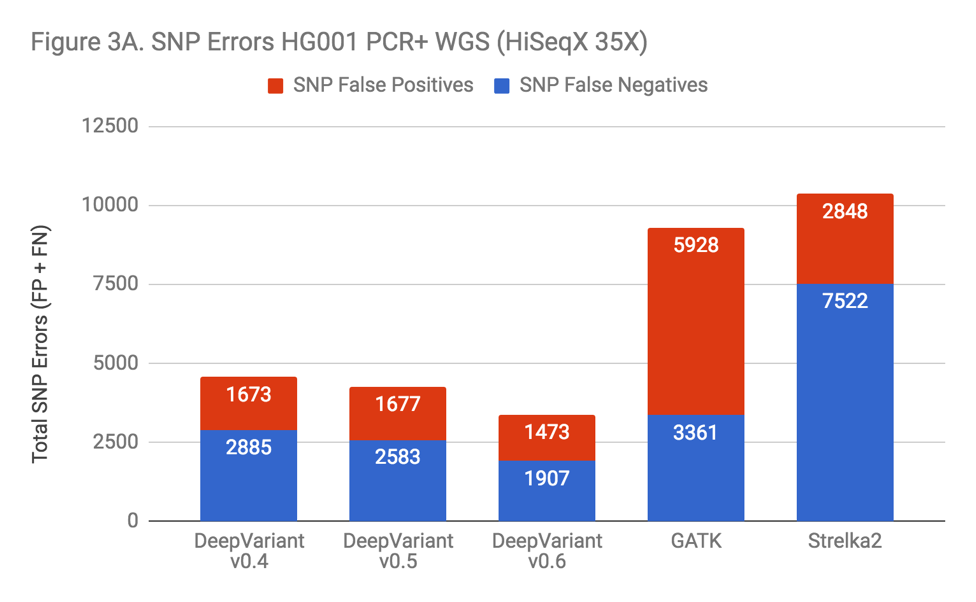
Indels in these samples were a problem for almost all of the callers. Indel error rates were so much higher in these samples that this is the dominant error mode across every method. The exception was Strelka2, which performed vastly better. Illumina clearly put great care into modeling PCR errors when developing Strelka2. Edico was also able to use this observation to improve their DRAGEN method, as detailed in our recent blog.
This observation was key to realizing that the performance penalty was due to PCR. When facing a problem with additional noise and complexity, it can be unclear whether the lack of signal makes the problem fundamentally less solvable or if the problem remains similarly tractable, but instead requires more effort. Strelka2’s performance was proof that current methods could improve significantly.
As Figure 3B shows, the inclusion of PCR+ data has a huge impact on the error rate of DeepVariant, with a 3-fold error reduction. Simply identifying the right training examples was sufficient to go from worse than GATK to 10% better than the prior leader, Strelka2.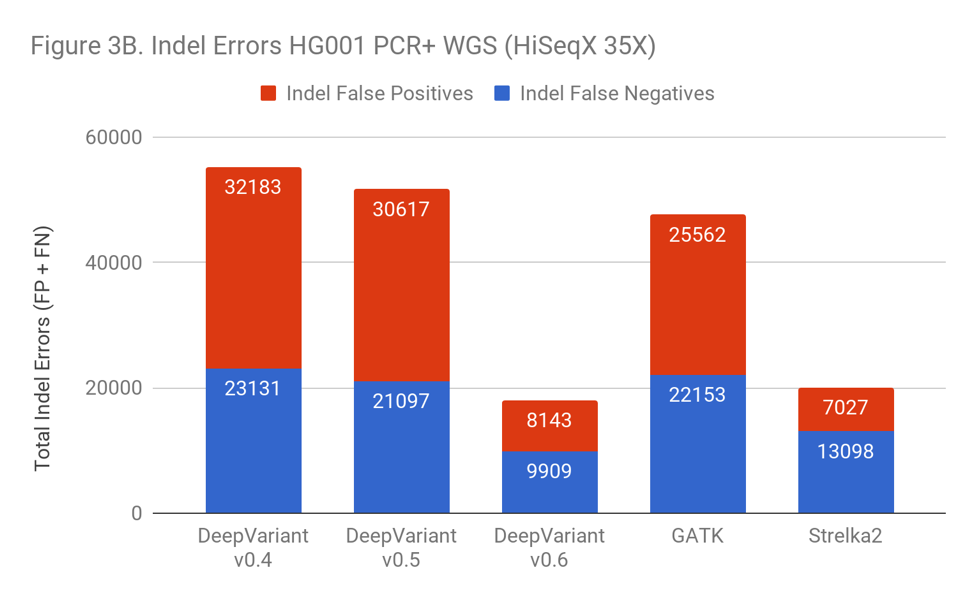
How Do New Training Data Impact Other Applications?
By including PCR+ and exome data in training, DeepVariant is now being asked to encode more information within its networks. Asking methods to multi-task in this way can lead to interesting effects and can force trade-offs which decrease overall performance.
An interesting phenomenon observed with deep learning methods is that challenging them with diverse, hard problems can sometimes improve general performance. Ryan Poplin, one of the authors of DeepVariant, discusses this phenomenon in a recent ML/AI Podcast on his work on an image classification method for diabetic retinopathy.
This could be understood in a few ways. First, harder problems may create pressure for network weights to identify subtle but meaningful connections in data. For example, a strong deep learning approach trained on novice chess opponents would only be so good. Another is that training examples that depend differentially on various aspects of a problem helps resist overfitting, allowing the model to discover and preserve subtle signals it could not otherwise. The result is a better generalist model.
There are many variables changed in the progression of DeepVariant from v0.4 to v0.6 – more overall training data has been added and tensors are changed – but if DeepVariant were to benefit from the complexity, one might hypothesize this would manifest in a more pronounced change on Indel performance, as that area is the most impacted in both the exome and PCR data. To test this, we ran each version on a “standard” benchmark – PCR-Free WGS HG002.
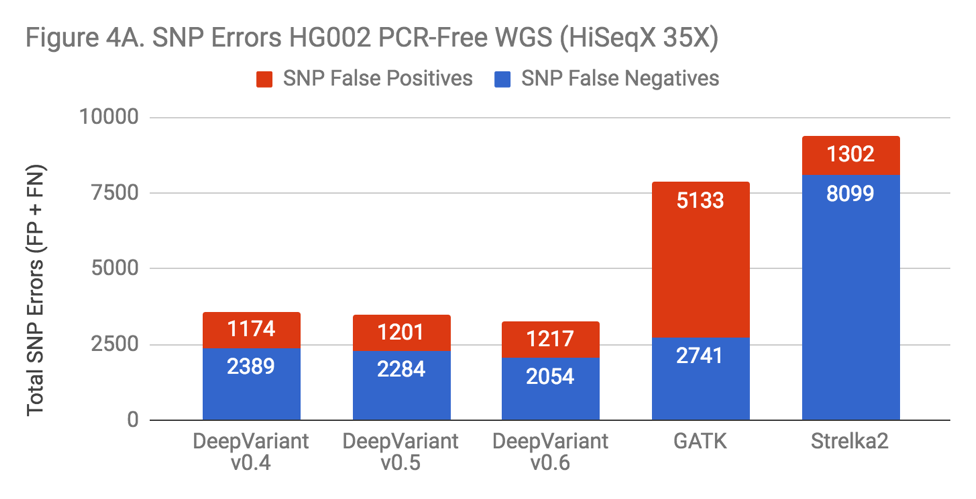
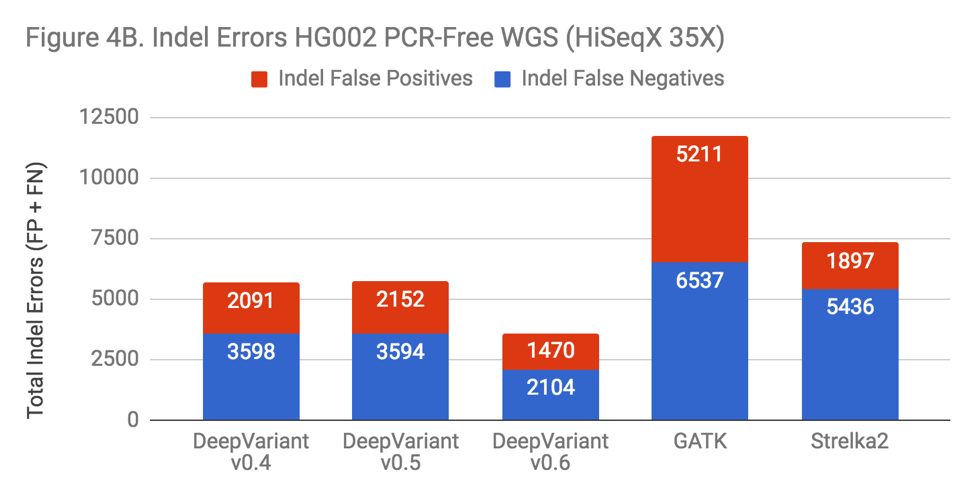
The new releases of DeepVariant show a trajectory of improvement, which is impressive given that DeepVariant led our benchmarks in accuracy even in its first release. SNP error rate is about 10% improved. The Indel error rate is improved more substantially, at a 40% reduction. Also, the greatest impact occurs with the addition of the PCR training data in v0.6. With this sort of uncontrolled experiment, we can’t conclusively say that this occurs because DeepVariant is learning something general about Indel errors from the PCR data. However, the prospect is enticing.
What Will Deep Learning Bring for the Field?
As deep learning methods begin to enter the domain of bioinformatics, it is natural to wonder how skills in the field will shift in response. Some may fear these methods will obsolete programming expertise or domain knowledge in genomics and bioinformatics. In theory, the raw methods to train DeepVariant on these new data types, confirm the improvement, and prevent regressions can be shockingly automatic.
However, to reach this point, domain experts had to understand the concepts of PCR and exome sequencing and identify their relevance to errors in the sequencing process. They had to understand where to get valid, labeled data of sufficient quantity and quality. They had to determine the best ways to represent the underlying data in a trainable format.
The importance of domain expertise – what is sequencing and what are its nuances – will only grow. How it manifests may shift from extracting features and hand-crafting weights to identifying examples that fully capture that nuance.
We also see these methods as enabling, not deprecating, programming expertise – these frameworks depend on large, complex training and evaluation infrastructures. The Google Brain team recently released Nucleus, a framework for training genomics models. Nucleus contains converters between genomics formats like BAM and VCF and TensorFlow. This may allow developers to tap into deep learning methods as specialized modules of a broader bioinformatics solution.
We hope that amongst the detail, this blog has communicated that deep learning is not a magic box. It remains essential for a scientist to carefully consider what the nuances of a problem are; how to rigorously evaluate performance; where blind spots in a method are; and which data will shine light on them.


.png)
.png)
.png)
{kind=link}