
Editor’s Note: This blog post was written by Don Freed, Bioinformatics Scientist at Sentieon. Email him at don.freed@sentieon.com.
Sentieon on DNAnexus provides clinicians and researchers an easy-to-use interface to process genomics data with state-of-the art tools in the cloud. In my previous post, we showed how the Sentieon Rapid DNAseq app is capable of processing an entire trio from FASTQ to VCF in about an hour. Today we’ll demonstrate the processing of targeted and exome-capture sequencing of tumor-normal paired samples.
Cancer is a genomic disease, arising as the result of sequential mutations in somatic cells. These mutations change the properties of cancer cells, leading to uncontrolled growth, cell death resistance, and eventually tumor formation and metastasis. Clinicians and researchers are very interested in digging into the genetic changes that give rise to cancer with next-generation sequence (NGS) data. NGS data allows researchers to better understanding the process of cancer and tumor formation and allows clinicians to determine the best course of treatment for cancer patients. As a result, the sequencing of paired tumor-normal samples is a common study design.
Somatic variant calling is a difficult task due to tumor heterogeneity and low abundance of tumor cells. Two of the more popular tools to address this task are MuTect and MuTect2 developed by the Broad Institute. High coverage across the protein coding sequences of the tumor genome is essential for the sensitive detection of low frequency somatic mosaic mutations. In targeted sequencing experiments, coverage of the targeted regions can exceed 1,000 or even 10,000 reads. Processing such high-coverage datasets is computationally intensive and by default MuTect (1.1.5) and MuTect2 (3.7) downsample the data to 1,000x and 500x, respectively, for improved runtime performance. Unfortunately, downsampling may also result in missed variant calls.
The Sentieon TNseq app on DNAnexus does not perform downsampling, enabling you to achieve the highest sensitivity for low frequency variants present in your sample. TNseq is also deterministic, given the same input data, the same variants will be called. In addition, the app produces results that match MuTect and MuTect2, but with a drastically improved runtime and cost savings.
Running TNseq Tumor-Normal Analysis on DNAnexus
For this case study, we used publicly available tumor-normal paired samples from SRP079186 and whole-genome sequence data from the Texas Cancer Research Biobank (TCRB). According to the project description in SRA, the tumor samples in SRP079186 consist of sequence data from small cell carcinoma mixed with squamous cell carcinoma of the esophagus, while the TCRB data are described in detail in this peer-reviewed publication. We obtained the FASTQ files from the European Nucleotide Archive and directly from the TCRB website and uploaded them into DNAnexus. To process the data, we used the Sentieon TNseq FASTQ to VCF app on DNAnexus. The pipelines produced VCF files of somatic variants called in the exome capture and targeted sequencing data. Alignment, duplicate marking, indel realignment, base-quality score recalibration, indel co-realignment and variant calling were performed with the DNAnexus workflows shown below.

Somatic variant calling on the targeted sequencing sample took only 37 minutes, while somatic variant calling from the paired whole-exome sequenced samples took 10 hours and 18 minutes. Variant calling in the 60x tumor and the 30x normal TCRB whole-genome sequence sample took 16 hours and 45 minutes.
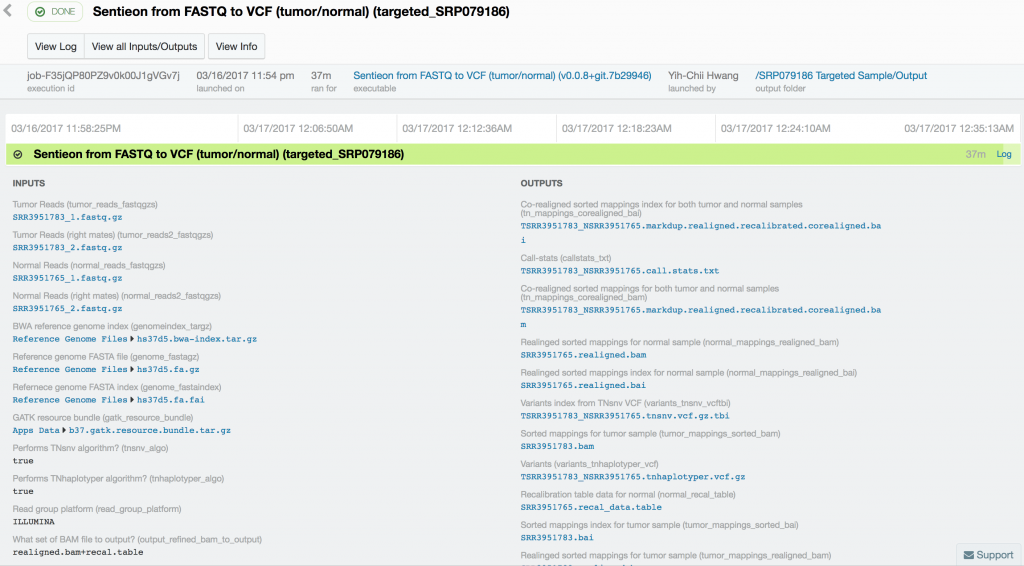
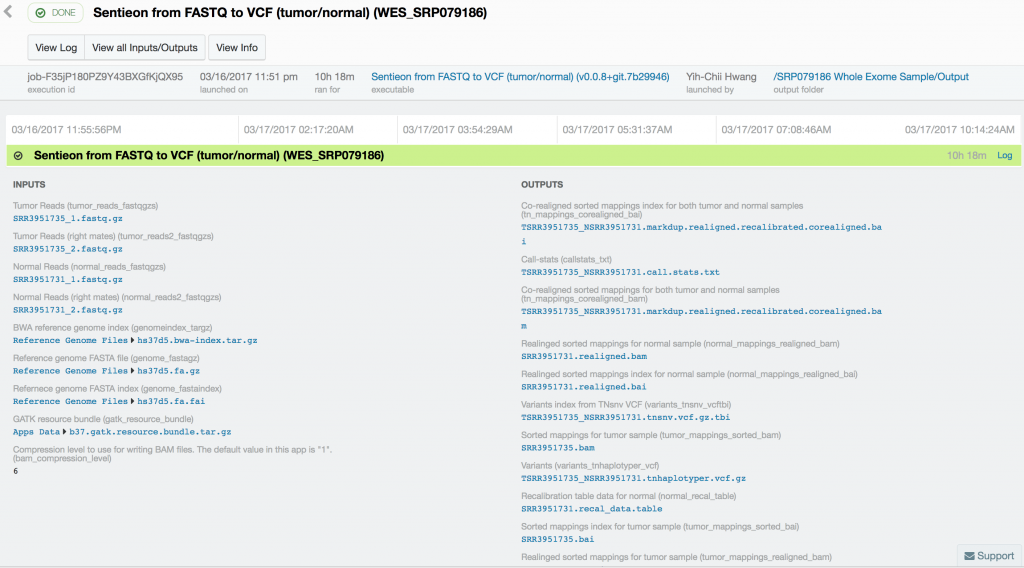
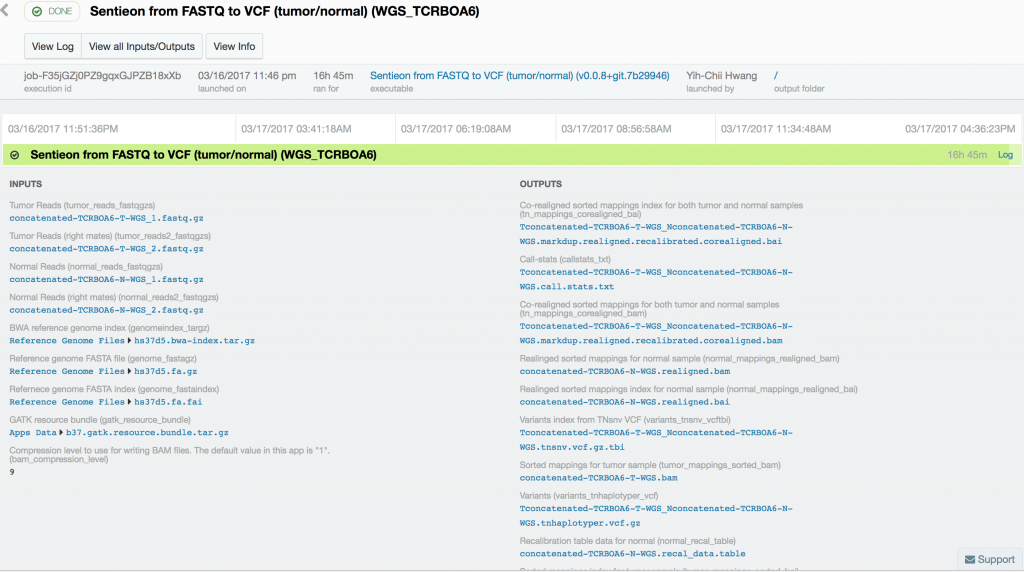
Using Sentieon’s TNseq FASTQ to VCF app, we were able to identify six and ten somatic mosaic mutations in the targeted sequence data, 381 and 739 somatic mosaic mutations in the whole-exome sequence data, and 5,812 and 2,171 somatic mutations in the whole-genome sequence data with TNsnv and TNhaplotyper, respectively. The number of non-silent mutations was 72 and 251 from the whole-exome sequence data as determined by TNsnv and TNhaplotyper, respectively. Other whole-exome sequencing studies of similar cancers have shown that 82 non-silent somatic mutations occur on average in these cancers. This is consistent with our findings given the differences in somatic variant calling pipelines used. Coverage of the targeted regions in the tumor sample exceeded 6,500x at some positions. MuTect2, with default settings, would have discarded more than 92% of the data at these sites, possibly resulting in false-negative or false-positive variant calls.
Digging deeper into the whole-exome data, we find p.R248Q and p.R158H somatic mutations in the gene TP53 encoding the protein p53, perhaps the best studied tumor suppressor protein. p53 functions as a key cell cycle regulator, stopping cell growth in the case of genomic damage. In many cancers p53 is inactivated as a method of bypassing cellular senesce. Germline inactivating mutations in TP53 cause a cancer predisposition syndrome called Li-Fraumeni syndrome. Somatic inactivating mutations in TP53 are expected in this sample; TP53 is the most frequent target of somatic mutation in esophageal squamous cell carcinomas and about half of all cancers have TP53 mutations. If you are interested in learning more about our analysis, you can see our workflow in the “Sentieon FASTQ to VCF (Tumor/Normal)” project on DNAnexus.
With the secure and collaborative cloud-based DNAnexus Platform and Sentieon TNseq FASTQ to VCF app, researchers and clinicians can rapidly and confidently identify somatic mutations from thousands of paired samples. Register here for a license-free trial of Sentieon tools on DNAnexus, available through April 7th.


.png)
.png)
.png)