
Editor’s Note: This blog post is written by Don Freed, Bioinformatics Scientist at Sentieon. Email him at don.freed@sentieon.com.
Introduction
At Sentieon we work hard to create the most efficient, accurate and robust tools for variant calling. Thanks to our partnership with DNAnexus, we are sharing the benefits of this hard work with you. Through April 7th, we are offering license-free access to Sentieon pipelines on DNAnexus, request access today to see how using Sentieon DNAseq you can obtain identical results to GATK at a fraction of the cost. In addition, Sentieon’s variant calling is deterministic; given identical input data, Sentieon will always call the same set of variants. Utilizing Sentieon’s tools on the DNAnexus Platform, clinicians and researchers can perform accurate and cost-effective analysis of petabyte-scale datasets with ease, seamlessly running analyses of an arbitrary number of samples simultaneously in the cloud.
Many of our customers use Sentieon tools to call variants from human samples. The typical Human genome contains some 4.5 million variants relative to the Human reference genome. While almost all of these variants are inherited, every individual has approximately 50 de novo variants, which occur uniquely in their genome. De novo variants are some of the most interesting genetic variants to study, they frequently cause rare sporadic diseases such as KBG syndrome, and have been implicated in complex disorders such as autism.
In this post, we’ll demonstrate the power of running Sentieon tools on DNAnexus by performing alignment with BWA, duplicate removal, base-quality score recalibration, indel realignment, haplotype-based variant calling and joint genotyping of a 30x whole-genome trio. Using these data, it is possible to identify de novo variants, the parental origin of some interesting inherited mutations, and examine the carrier status of this individual for rare recessive mutations. With the Rapid DNAseq app on DNAnexus, processing an entire trio takes about an hour. Whether you have a cohort of three or 3,000, by leveraging the power of the DNAnexus Platform and the scalability of the cloud, any size cohort can be processed incredibly fast.
Running analyses on DNAnexus
For this trio analysis, we used data from the Illumina Platinum Genomes dataset for individuals NA12878, NA12891, and NA12892 downsampled to 30x. The original fastq files can be found at the European Nucleotide Archive. To process the data, we used the Sentieon rapid DNAseq app on DNAnexus. We called variants in GVCF mode and input the gVCF files into the Sentieon GVCFtyper resulting in a single multi-sample VCF file for the entire trio. We easily accomplished this by using the DNAnexus workflow shown below.
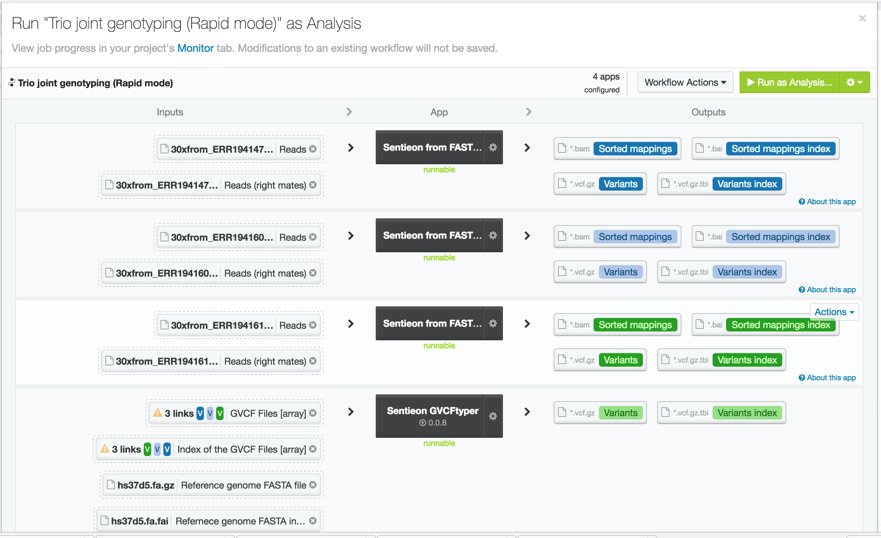
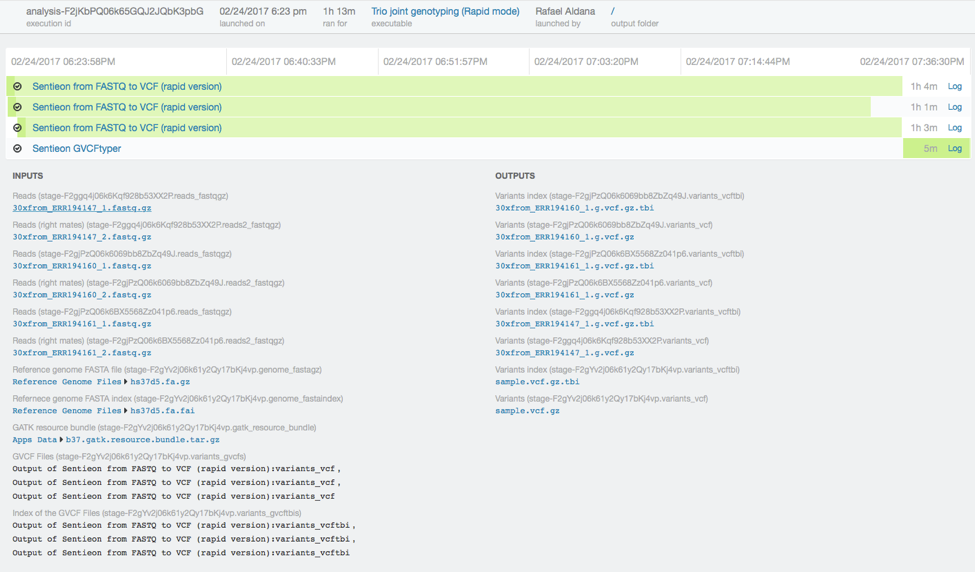
In total the analysis took just 73 minutes.
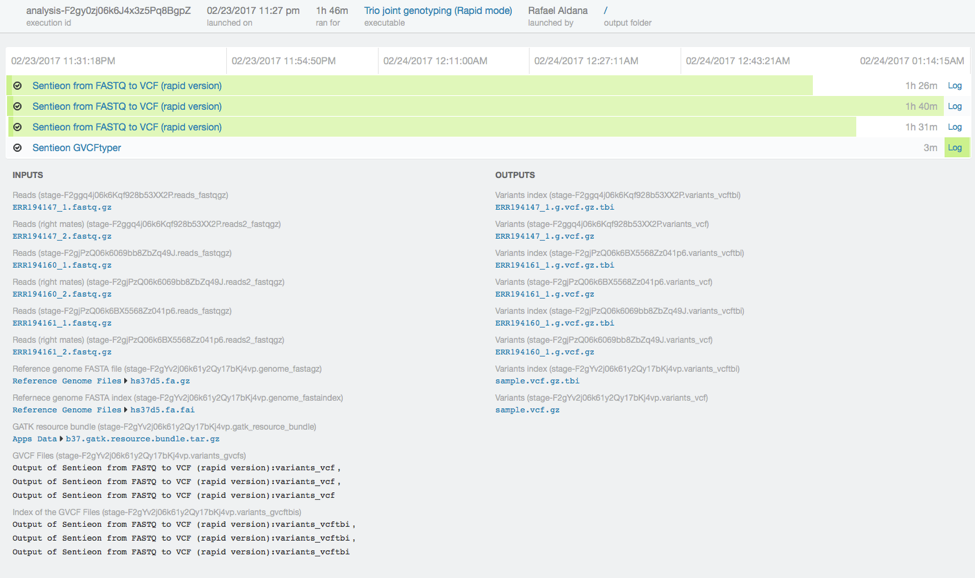
We performed the same analysis with the original 50x dataset in one hour and 46 minutes. Runtimes scale approximately linearly to the input coverage.
We identified 2,458 de novo mutations in NA12878, well above the expected 50, although this increase has been previously attributed to primary cell somatic mutations or mutations introduced during immortalization and subsequent passage of the sequenced cells. We can see that NA12878 is heterozygous for both rs2472297 and rs6968865, which have been associated with increased coffee consumption.
Utilizing the DNAnexus cloud-based platform and Sentieon tools, our rapid DNAseq and joint genotyping runtimes easily scale to thousands of samples. You can view everything we ran in this public project: Rapid trio genotyping.
Register here for a free trial of the rapid DNAseq tool.




