

Crumble is a newly published NGS compression method by James Bonfield, Shane McCarthy, Richard Durbin of the Sanger Institute. In this post, we demonstrate that Crumble provides size savings of 20-70% for BAM and CRAM files from HiSeq2500, HiSeqX, and NovaSeq Whole Genome Sequencing (WGS) and exomes. We profile the resulting variant calls, showing generally similar results when using Crumble, even slightly improving variant-calling accuracy on the same sequencing data.
Introduction
Among computational fields, genomics is especially data-heavy. For a 30x WGS sample, the size of the BAM file can easily exceed 100 GB. The collective size of genomics projects is growing at a tremendous rate. One highly cited work by Stephens et al. projects that, with current trends, the volume of NGS data in 2025 will exceed the data footprint of YouTube, Twitter, and astronomy.
Most of the data footprint is in BAM files. In this format, the data footprint is divided between Read Names, the Sequence itself, Quality Values (QV) and Tags. The CRAM format uses lossless reference compression for the sequence, pushing the relative data footprint toward storing the quality values (see our Readshift blog for background on QV).
 In an uncompressed (SAM) file, each quality value occupies a bin with a fixed amount of space (see the figure on the right, where each quality value is represented as a different shape). The BAM format compresses this data with a standard method similar to gzip. To simplify compression methods greatly, they identify recurring patterns within the data that can be used to pack elements together. More irregular data (data with higher entropy) is harder to compress. This blog from Dropbox contains interesting examples on how compression methods can work.
In an uncompressed (SAM) file, each quality value occupies a bin with a fixed amount of space (see the figure on the right, where each quality value is represented as a different shape). The BAM format compresses this data with a standard method similar to gzip. To simplify compression methods greatly, they identify recurring patterns within the data that can be used to pack elements together. More irregular data (data with higher entropy) is harder to compress. This blog from Dropbox contains interesting examples on how compression methods can work.
As Illumina has progressively increased instrument throughput, it has fought the growing friction in managing data footprint by decreasing the range of possible QV (HiSeq2500 – 40, HiSeqX – 8, NovaSeq – 4). The figures below illustrate how packing a stream of QV pieces together is easier when there are fewer types of QVs and these types are more similar. These changes are generally synergistic with other advanced compression technologies such as Petagene.
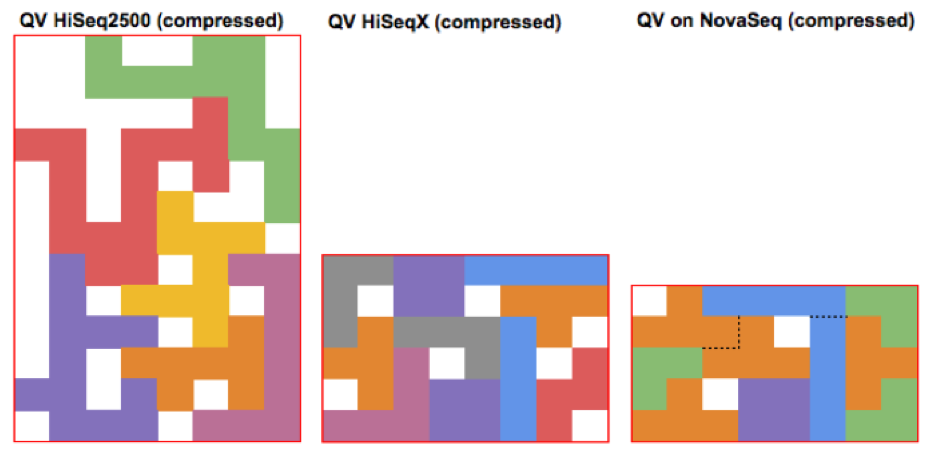
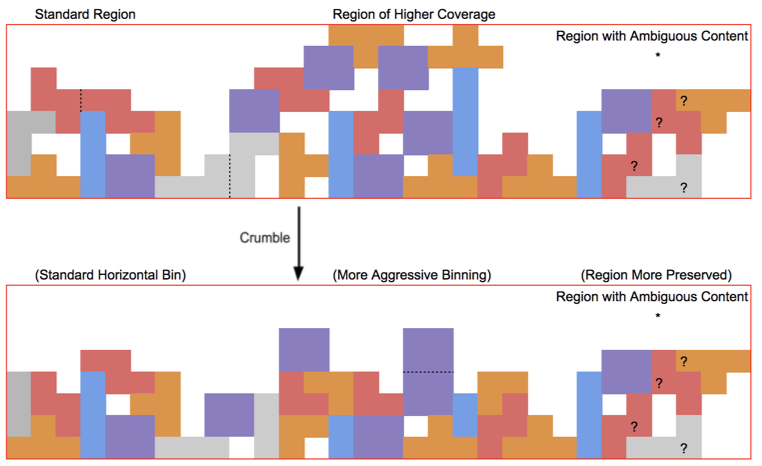
The Crumble paper calls this strategy “horizontal” because it applies generally to all bases. Crumble additionally applies a “vertical” approach to consider the context of surrounding reads, preserving more quality values in regions of lower coverage or in signals of interest (like variant positions) and compressing more in regions where reducing entropy is unlikely to impact results. The figure below attempts to conceptually represent some of the conditions under which Crumble bins more or less aggressively.
Results: Crumble Saves Significant Space for Both Whole Genomes and Exomes
To assess Crumble, we first applied it to several WGS and exome BAM and CRAM files produced across different machines. As Figure 1 illustrates, Crumble saves significant space across all types of WGS files, with larger savings in HiSeqX compared to NovaSeq. In these charts, Crumble is run at its default settings. Lower numbers are better (i.e. BAM/CRAM which takes less space).
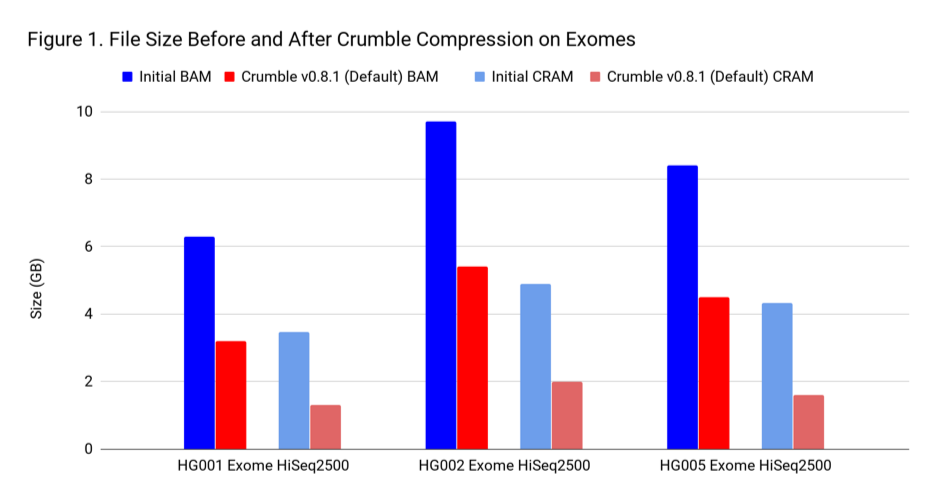
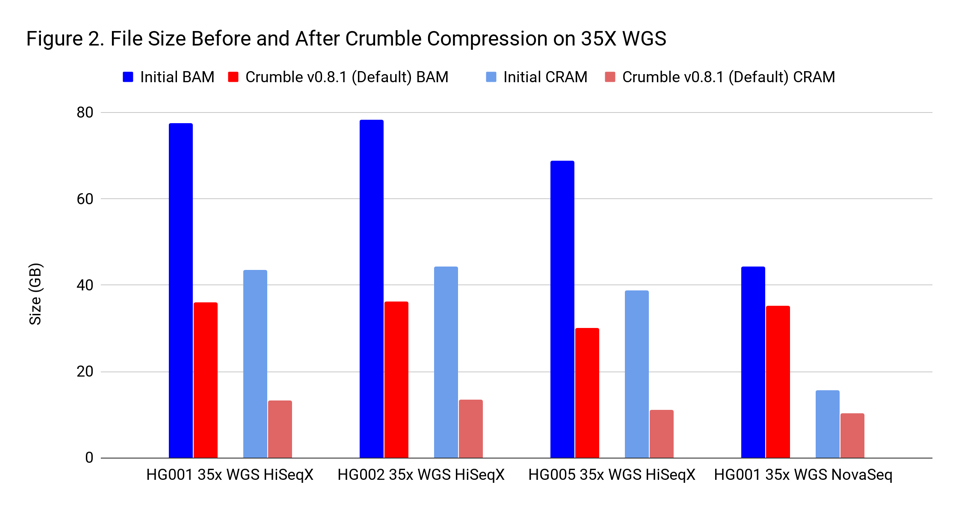
The average savings in size relative to the starting state are:
| Sample Type | Percent Size Savings (BAM) | Percent Size Savings (CRAM) |
| WGS HiSeqX | 55 % | 70 % |
| WGS NovaSeq | 20 % | 34 % |
| Exome | 47 % | 60 % |
Applying Crumble Improves Variant Calling Results in Almost All Conditions
Crumble’s intelligent application of binning allows it to minimize possible negative consequences. Interestingly, Crumble has a positive impact on variant calling results overall. We suppose this occurs because Crumble bins more aggressively in regions of unusually high coverage or poor mapping. Though QVs indicate the probability that a given base is in error, taking into account the additional probability that a read is mismapped is a more complex calculation that variant callers are likely overconfident about. Crumble’s binning may correct for this by reducing unfounded confidence.
Crumble improved accuracy on NovaSeq WGS for almost every pipeline, with substantial decreases seen in several. In all charts that follow, lower values on the y-axis (that is fewer errors) is better.


For exomes, the changes in accuracy were less pronounced. Only GATK showing a substantial difference, with an improvement of 10%.
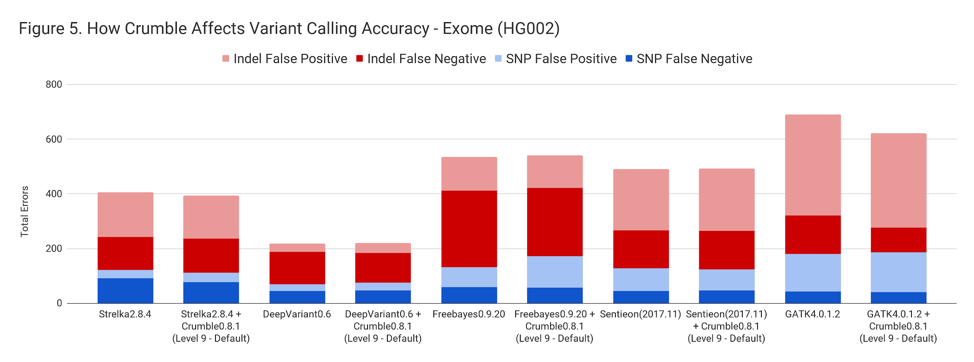
The following table summarizes how Crumble impacts variant calling across the use cases. In this, larger negative numbers indicate that Crumble has a larger improvement. In almost every case, Crumble improves results in 73% of investigated cases, with substantial (~10% of more) improvements in 33 % of cases. In those cases where Crumble made pipelines worse, the impact was small. GATK4, in particular, was consistently helped by Crumble.
Change in total errors by using Crumble v0.8.1 relative to baseline (non-Crumble).
| Strelka2 | DeepVariant | Freebayes | Sentieon | GATK4 | |
| HiSeqX WGS | – 1.3 % | – 19.2 % | – 5.2% | + 3.1% | – 27.1% |
| NovaSeq WGS | – 1.2 % | + 3.5 % | – 2.3% | – 10.3% | – 22.6% |
| Exome | – 3.0 % | + 1.4 % | + 1.3 % | + 0.1 % | – 9.9% |
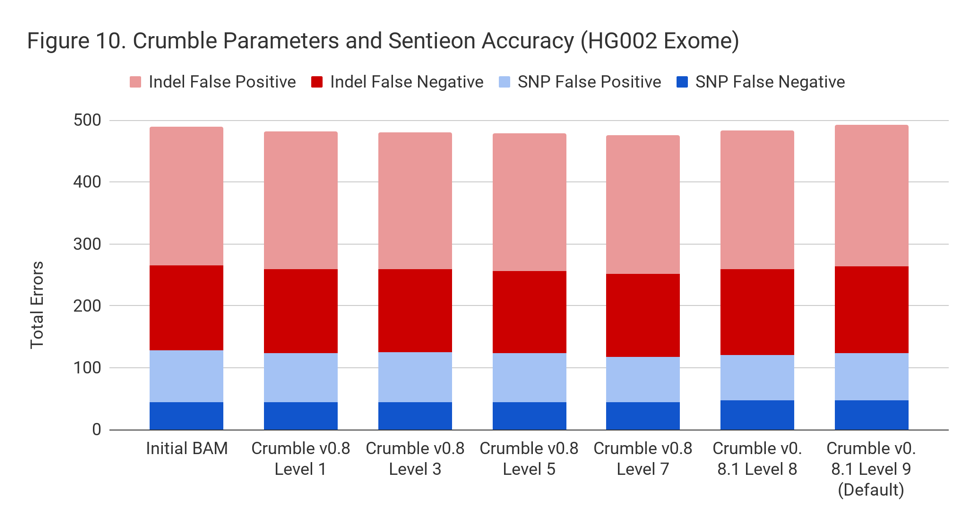
Crumble Level Parameter is More Important for Exomes. Using Level 9 (Default) is Different for Both WGS and Exomes
Crumble comes with presets for parameter combinations that correspond to increasingly aggressive binning strategies (Level 1, 3, 5, 7, 8, and 9, with 9 as the default). We applied each of these settings to 4 WGS samples (HiSeqX HG001, HG002, HG005, NovaSeq HG001) and 3 exomes. In every case, our results were consistent between WGS samples and between exome samples. Therefore, we present results with only one representative in each.
Our initial investigation of level used Crumble v0.8. Version 0.8.1 was released 2 weeks after, adding a new Level 9 and changing the old Level 9 to Level 8, but not otherwise changing settings. So we have only re-run the new modes in this chart.
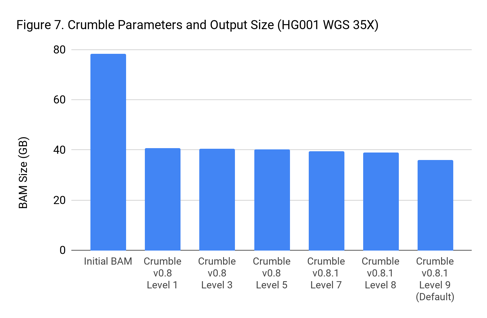
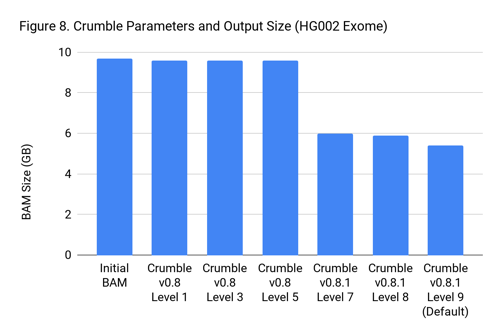
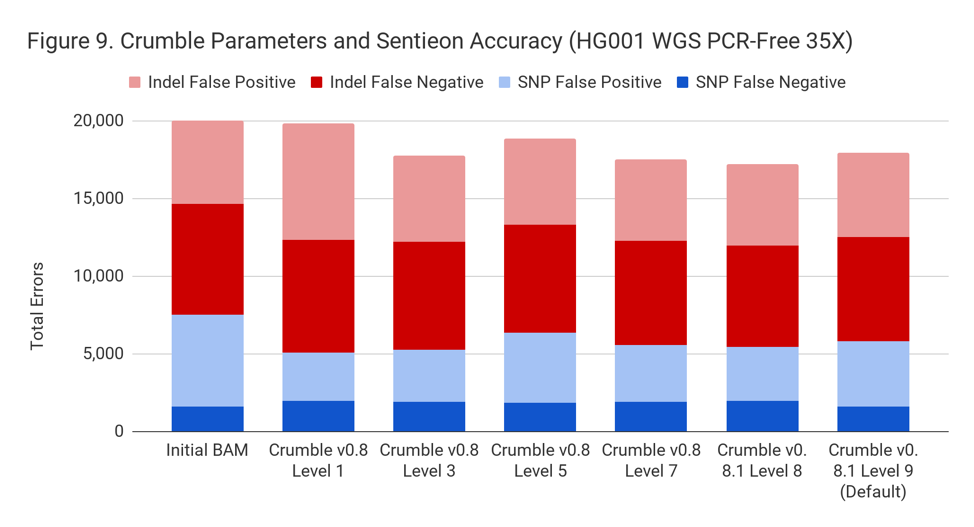
Crumble Is Fast and Cheap to Run
By running Crumble, a user chooses to pay a compute cost in order to achieve persistent savings in storage. This cost-benefit decision depends on the speed of the method. Fortunately, Crumble is extremely fast, especially when compared to methods for mapping samples or calling variants. We are generally able to run Crumble on a 35X WGS in just 8 CPU-hours.
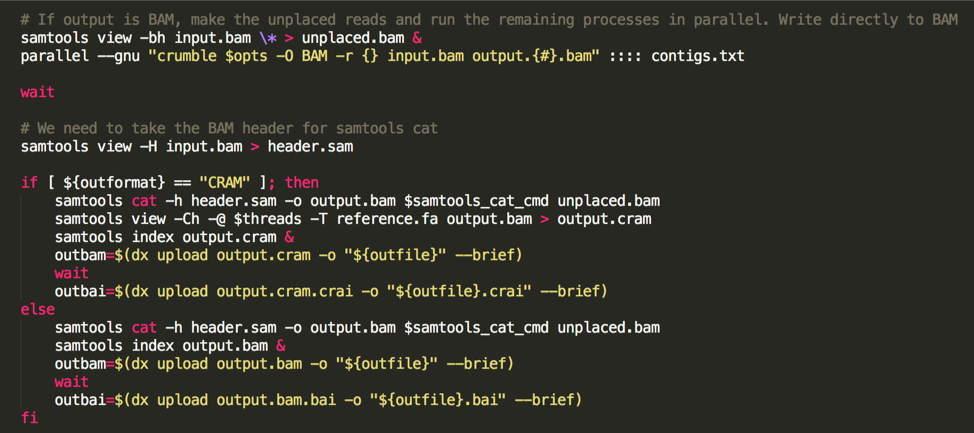
Unfortunately, Crumble does not currently support multi-threading. However, users can restrict it to running on a genomic region. So, to run Crumble quickly and efficiently, we do the following:
- Generate a contig list from the BAM header,
- Set the unplaced reads aside
- Use gnu-parallel to apply Crumble in parallel to each chromosome.
- use samtools cat to combine everything.
(the following image is a code snippet from our app code):
With this, we can execute the Crumble program in about 1 hour of wall-clock time on an 8-core machine (download and uploading to the cloud worker both take a bit of additional time).
The break-even point at which point the storage savings justify the extra compute cost depends on your relative costs of each. However, under most realistic scenarios, running Crumble will pay back the investment in just a few weeks to months, and will accrue lasting savings well beyond that. There are other benefits to reducing data size: it is easier for collaborators to access, and it takes less time to load into and out of cloud workers from cloud storage.
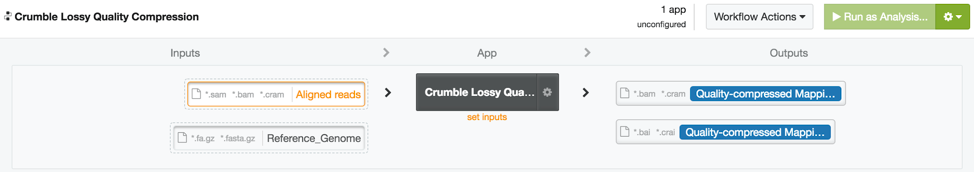
Crumble on DNAnexus
An efficiently wrapped app to run Crumble is available to all users on the DNAnexus app library at https://platform.dnanexus.com/app/crumble (platform login required to access this link). This app can both take data and produce output in BAM or CRAM output (CRAM output requires a matching reference genome).
Final Notes
These tests are all performed on germline sequencing. It is unknown how Crumble would interact with somatic methods. There are other operations which can shrink BAM/CRAM files further without impact, such as lossy compression of read names. We hope to explore these in a later post.
Conclusion
We hope we have demonstrated that Crumble is a useful tool capable of achieving significant space savings on diverse data. Under most conditions, Crumble has a negligible or positive impact of downstream processes. It can be run at Level 7 to effectively compress both exomes and WGS. The rapid speed of Crumble means that it is favorable to run from a cost-benefit evaluation.
As genomics continues to grow in scale, we will need methods such as Crumble that enable us to consider how to ensure that, as a field, we have solutions which can scale to future needs. The value of genomics should be judged not by the magnitude of its data footprint, but by the value the field brings to society.
We thank the authors of Crumble, James Bonfield, Shane McCarthy, and Richard Durbin of the Sanger Institute, for their hard work in making this tool available to the community, and for providing helpful comments and suggestions on this blog.


.png)
.png)
.png)