

In this blog post, we explore variability in the quality of sequencing runs. We present a method which allows the generation of benchmarking sets of real sequence data across arbitrary quality ranges. Finally, we present benchmarks of popular variant calling pipelines across these diverse quality sets.
Introduction
The recent generation of gold-standard genome datasets – such as the Genome in a Bottle (GIAB) and Platinum Genomes – has given greater understanding about the quality of sequencing. Competitions held on PrecisionFDA have driven the innovation and refinement of new methods. Sentieon, Edico, and DeepVariant, each a winner of one or more of Consistency, Truth, and Hidden Treasures Challenges, have proven the power of standards to drive innovation. These gold-standard datasets used to construct truth sets and challenges were produced with great care and attention to quality.
We wondered how directly these sets reflect common, real-world sequencing data. “Quality” of sequencing is complex – factors such as library complexity, coverage, contamination, index hopping, PCR errors, and insert size distribution are all important. For our exploration, we use a quantifiable, unambiguous, and straightforward measure – the sequencer-reported base quality.
FASTQ base qualities estimate the probability that a base call (A, T, G, or C) is in error. These errors make reads more difficult to correctly map and produce noise that variant callers must contend with. Monitoring of base qualities is often a component in QC.
We score each pair of forward and reverse reads with the expected value for the number of errors present in the pair. The combination of these values for each read pair in a sequencing run creates a distribution with a mean and standard deviation for errors.
Comparing GIAB, PrecisionFDA Sets to General Data
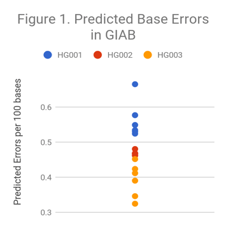
First, we investigate the Illumina HiSeq2500 contributed to GIAB. These data, along with contributions of PacBio, Complete, 10X, BioNano, Oxford Nanopore, and Ion Torrent were used to create the GIAB truth sets.
As Figure 1 indicates, there is some variability in quality – HG003 samples have about 30% less of an error rate than HG001. However, overall base accuracy is high.
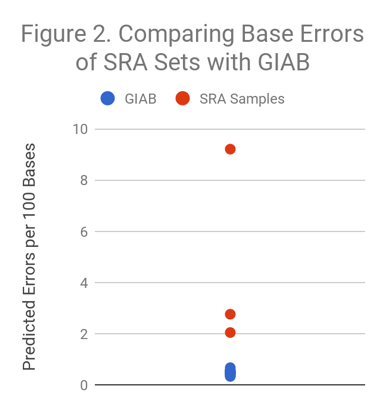 We selected 3 readily available HiSeq2000/HiSeq2500 sets from SRA and performed the same calculation. As Figure 2 illustrates, other datasets range from modestly higher (~3x) to dramatically higher (~20x) error rates. The blue dots in this figure are the same as all of the dots in Figure 1.
We selected 3 readily available HiSeq2000/HiSeq2500 sets from SRA and performed the same calculation. As Figure 2 illustrates, other datasets range from modestly higher (~3x) to dramatically higher (~20x) error rates. The blue dots in this figure are the same as all of the dots in Figure 1.
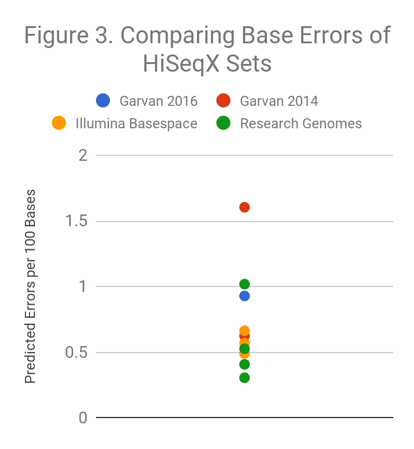
We applied a similar investigation to datasets from the HiSeqX. In Figure 3, we compare predicted error rates for the 2016 Garvan WGS dataset contributed to the PrecisionFDA Truth challenge, several HiSeqX replicates produced by Illumina and available on BaseSpace, several HiSeqX runs performed by Academic Medical Centers, and the first publicly available 2014 HiSeqX run from Garvan that we noted as difficult for Indel calling in a prior blog post.
With these HiSeqX sets, there is a 5-fold variability in predicted error rate between the highest and the lowest samples. How do differences of this magnitude relate to the quality of variant calls from a sample? Answering this question is not easy since the diversity of datasets are not on the genomes (HG001, HG002, or HG005) that have truth sets for. To address this, we have developed a method called Readshift, which allows us to resample existing runs to create real datasets of diverse read quality.
Readshift
Readshift uses biased downsampling from a dataset of larger coverage to create a shifted quality distribution at lower coverage. (See Figure 4 for a conceptual example.)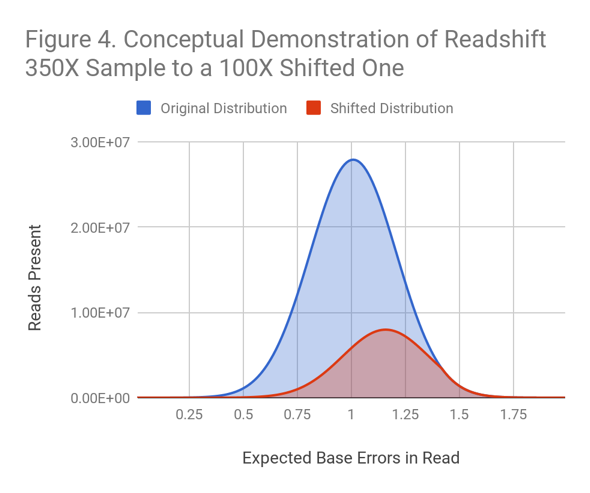
It does so by modeling the original and final samples as normally distributed and calculating what the probability for inclusion of each read would be required to reach the desired distribution.
Readshift does not force the final distribution to be normally distributed. Rather, it acts as a transformation which allows shifting of a distribution in a way that preserves the shape of the original distribution.
Importantly, the data produced by Readshift is not simulated – these are real reads produced by a real sequencer and thus carry all of the intricacies, biases, and idiosyncrasies associated with real data. No individual base or quality value is altered.
All of the code for Readshift is released with an open source license on Github – https://github.com/dnanexus/Readshift. The FASTQ data in this investigation are available in a public DNAnexus project for download or general use, as are the Readshift apps.
Shifting HiSeq2500, HiSeqX, and NovaSeq Datasets
Several high-coverage datasets exist for HG001 on the HiSeq2500, HiSeqX, and NovaSeq platforms. We took 200X HiSeq2500 data submitted for GIAB, 200X HiSeqX data available on BaseSpace, and 350X Novaseq data available on BaseSpace and applied Readshift to increase the predicted errors in reads by 0.5, 1.0, 1.5, and 2.0 standard deviations relative to the prior mean. To confirm that no artifacts are introduced by the shift machinery, we also applied Readshift with a shift of +0.0 standard deviations.
This range roughly covers the difference between the best and worst HiSeqX sets shown above. The magnitude of the possible shift is determined by the amount of starting coverage – higher coverage allows larger shifts without clipping the distribution.
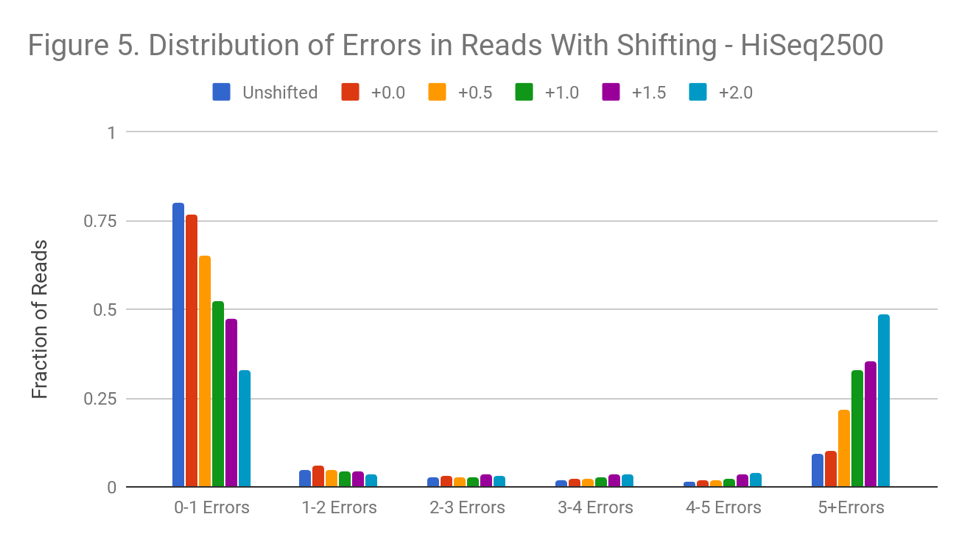
Figure 5 shows the distribution of read errors after the shift. Though we use the probability density function of the normal to do the transformation, shifting qualitatively preserves the distribution shape. Both original and final distributions are left-leaning curves. At the higher degrees of shift, much of the decline in read quality appears driven by reads with a strong decline in quality toward the 3’ end, or reads with a bad mate.
Evaluating WGS Pipelines on Noisy Datasets
We evaluate several popular WGS variant calling pipelines on this shifted data starting from 35X coverage FASTQ files. All analyses are done against the hg38 reference genome. Accuracy is assessed on the confident regions using the Genome in a Bottle Truth Sets, with the evaluation performed using the same method that we use in PrecisionFDA.
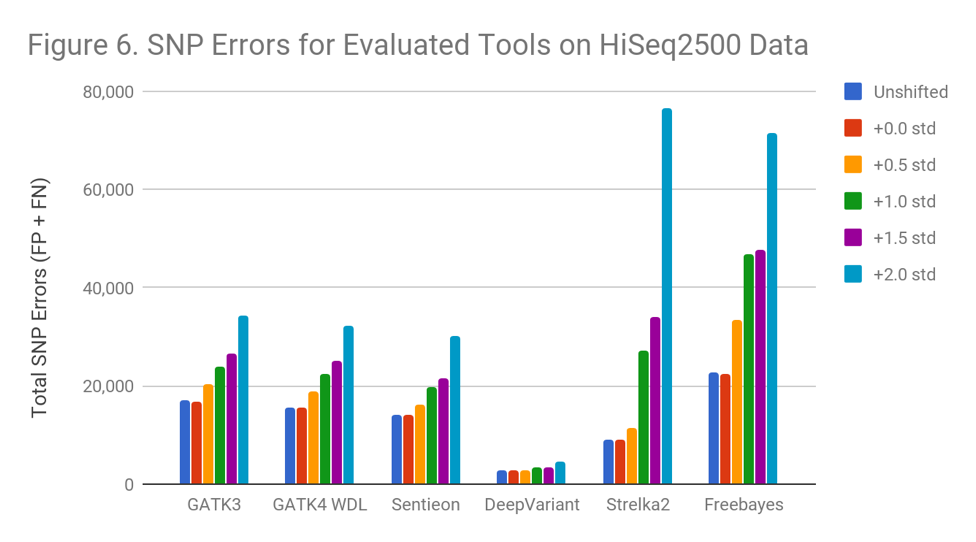
Figure 6 demonstrates that tools behave differently when used to call increasingly noisy data . Freebayes begins to suffer immediately, while Strelka2 suffers once the shift exceeds 0.5 standard deviations. Even the most resistant programs perform significantly worse (GATK 100%, DeepVariant 68% more errors) at a shift of 2.0 standard deviations. Sentieon avoids downsampling to achieve reproducibility, but otherwise matches GATK3.
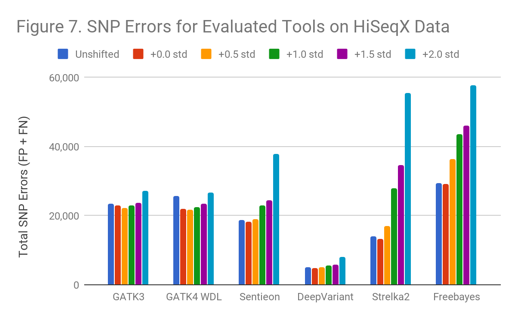
 The SNP results for HiSeqX follow a similar pattern as indicated in Figure 7.
The SNP results for HiSeqX follow a similar pattern as indicated in Figure 7.
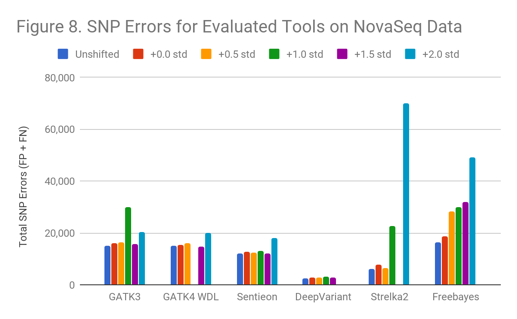
For NovaSeq (Figure 8), though the data is qualitatively similar to the HiSeq2500 and HiSeqX runs, other complexities occur. Generally, low-quality runs took significantly longer to finish for this data. Some programs either failed or produced results that suggest they did not call several regions for high coverage. The NovaSeq uses 2-color chemistry which does not enable it to indicate an unknown base as N. This may make reads with more errors hard to uniquely map, causing an increase in secondary alignments and regions of unmanageable coverage.
This suggests that filtering of NovaSeq data may be more important than with previous sequencers in low-quality runs. (GATK3+1.0, GATK4+1.0, Strelka+1.5, and DeepVariant+2.0 were impacted).
Indels
The results with Indel calling was different than those for SNPs. Apart from Freebayes, none of the variant callers suffer an accuracy penalty on the lower-quality data.
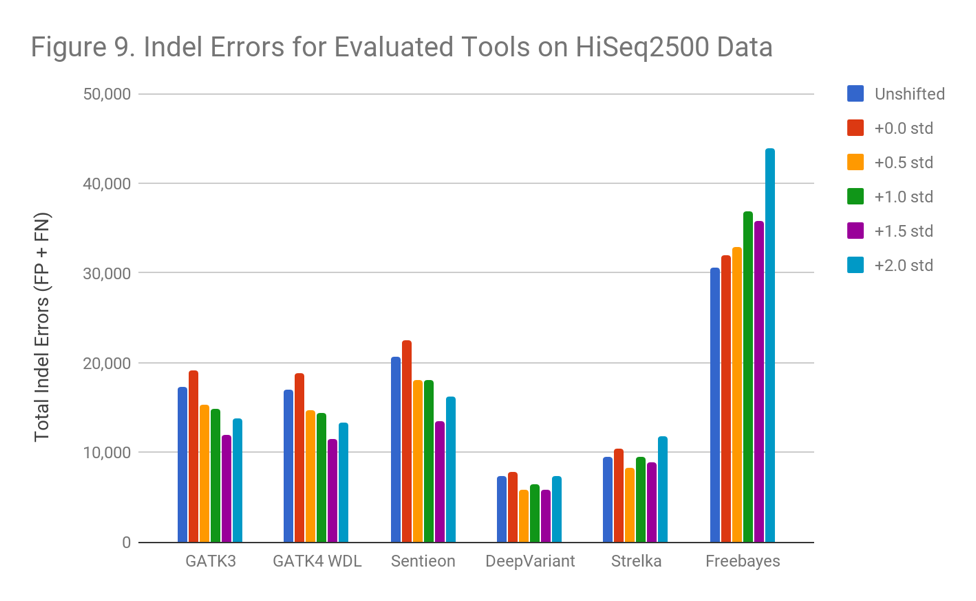
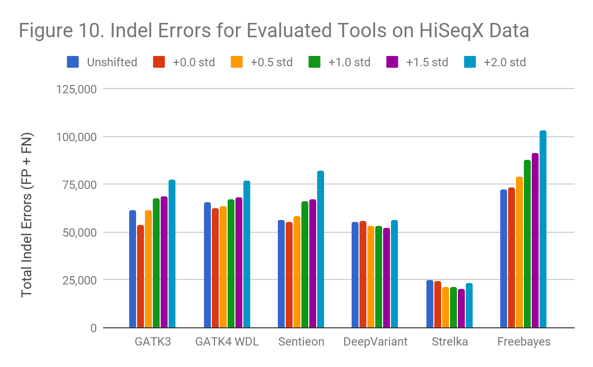 In the HiSeqX data (Figure 10), the Indel error rate is much higher even in the original data. This is the same phenomenon we observed in the early 2014 Garvan HiSeqX run we found in a prior blog post; however, these are different runs. It is possible that many early HiSeqX runs have this issue, or that some other aspect of the preparation is at fault.
In the HiSeqX data (Figure 10), the Indel error rate is much higher even in the original data. This is the same phenomenon we observed in the early 2014 Garvan HiSeqX run we found in a prior blog post; however, these are different runs. It is possible that many early HiSeqX runs have this issue, or that some other aspect of the preparation is at fault.
Interestingly, though Indel callers arerobust against these extreme shifts in read quality, they suffer strongly from whatever occurred in the HiSeqX runs.
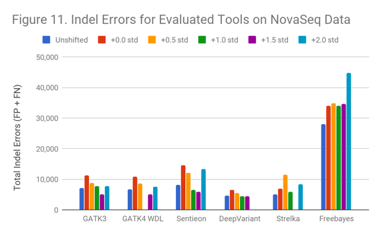
The NovaSeq data (Figure 11) is qualitatively similar to the HiSeq2500 results.
The fact that the callers exhibit greater robustness against Indel errors could be an opportunity to improve SNP calling. Although the fact that the definition of base quality values map more directly to SNP events than Indels, reads in the higher shifts often have long stretches of low-quality bases toward the 3’ end. Based on this, it seems likely that something in the way tools handle indels mitigates the low-quality reads. This could be a heavier reliance on haplotype construction, Indel realignment, or local assembly to resolve Indels. It could also be that since Indels span multiple bases, there is a greater signal to distinguish random errors from meaningful differences. If this is the case, this ability to gain nearby corroborating evidence might be achievable by phasing nearby variants or looking whether nearby potential errors co-vary.
Increased Computational Burden on Low Quality Sets
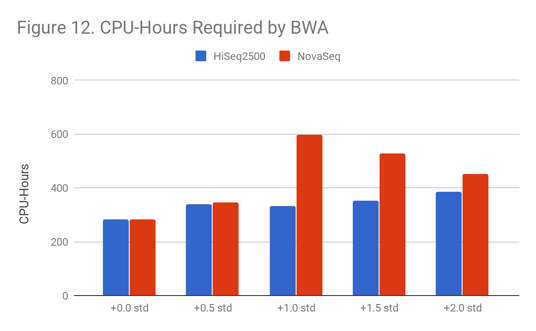 As Figure 12 and Figure 13 indicate, lower quality samples carry a higher computational burden. For BWA, reads become harder to uniquely map, which also causes an increase in secondary alignments.
As Figure 12 and Figure 13 indicate, lower quality samples carry a higher computational burden. For BWA, reads become harder to uniquely map, which also causes an increase in secondary alignments.
This problem is more severe for NovaSeq data, perhaps because 2-color chemistry lacks a way to designate a base as “N” (“no color” corresponds to “G”) for NovaSeq.
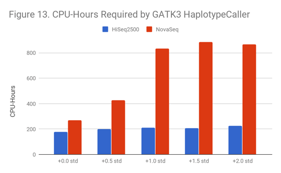 For GATK3 HaplotypeCaller, this can cause samples to require 3x as much compute, even at moderate shifts. We believe that GATK3 is affected because secondary align reads pile up in poly-G/poly-C regions of the genome, and this generates a large number of candidate haplotypes to consider.
For GATK3 HaplotypeCaller, this can cause samples to require 3x as much compute, even at moderate shifts. We believe that GATK3 is affected because secondary align reads pile up in poly-G/poly-C regions of the genome, and this generates a large number of candidate haplotypes to consider.
We think that many programs may suffer similar impacts, particularly any process which considers across candidate events at low confidence (e.g. many possible haplotypes) and selects the best choices. This may be worse for methods in somatic calling, or detection of events in cell-free DNA. Even in pristine reads with a 3/1000 error rate, any given mismatch is 3x more likely to be a sequencing error than the human variation rate. Germline callers manage this because the signal from germline events is consistent. Somatic callers must consider candidates at low frequencies or risk losing rare, but impactful, events. We hope to discuss this and strategies for mitigation in a future post. Note that the strong majority of NovaSeq runs are just fine – and standard read filtering tools seem to work for those that have issues.
Conclusion
By providing this analysis, we hope to highlight the fact that not all sequencing runs are the same and that this is important to their analysis, the choice of tools for them, and how we interpret tool evaluations on gold standard data. By releasing both the code https://github.com/dnanexus/Readshift to perform the shift and the data (https://platform.dnanexus.com/projects/F9K5zYQ0ZQBb5jJf6X2zZPv1/data/), we aim to help the community improve the accuracy and robustness of their methods. Readshift can be applied to any emerging benchmark as long as high coverage sequencing exists, such as the CHM1-CHM13 dataset developed by Heng Li et al.
This work raises a number of additional areas we have begun to investigate, including the best strategies to filter reads to protect against low-quality sets, the differences in impact on precision versus recall across tools and sequencing platforms, and the potential for ensembling analytical methods to shield against outlier sequencing runs.
If you are interested in collaborating with us on this work, we want to engage scientific partners. If you have sequence datasets on NA12878/HG001, HG002, or HG005, particularly from runs that turned out to be bad, and want to help us use them to make better benchmarking resources: Readshift only gets stronger with more data. Please reach out to us at readshift@dnanexus.com.


.png)
.png)
.png)