
Current sequencing technology and computational algorithms support the construction of phased diploid genome assemblies, and these are useful for studying genomic regions with high variability. For repeat-rich regions, mapping based methods with short reads usually fails to give reliable variants call and it is even harder to get a phased variant callset.
A recent study, published in Nature Communications, describes efforts to develop the first variant benchmark using a diploid assembly, overcoming the obstacles of the mapping only approaches, using the HG002 dataset from the Genome in a Bottle (GIAB) consortium. Below is a summary of the findings from the recent collaboration between DNAnexus and GIAB, which builds on the work from an NCBI pangenomics hackathon hosted by UCSC in March of 2019 and on GIAB’s existing efforts to develop truth datasets that support genomic research.
The research team specifically focused on benchmarking variants in one of the most polymorphic and medically important parts of the genome – the major histocompatibility complex (MHC). The MHC plays a crucial role in adaptive and innate immunity among other activities. There is a high level of variability between individual genomes that makes characterizing this region particularly challenging. Previous benchmarks that relied primarily on short-read mapping methods are unable to map large portions of the region’s sequences because of large differences between the reads and the reference. This is particularly true for very repetitive regions of the genome that contain things like segmental duplications and tandem repeats.
Recent improvements in de novo assembly using more accurate, consensus long reads have made it possible to “represent both haplotypes without suffering from small indel errors due to error-prone long reads,” the researchers note in the paper. Previously published assemblies still had “substantial error rates for small variants of at least 10 % due to their reliance on error-prone long reads, and the individual long and ultralong read assembly incompletely resolved haplotypes.” Linked read assembly methods have also been helpful for resolving the MHC but the results are “fragmented” with similar error rates for small variants.
To generate this benchmark, the researchers developed a local de novo assembly approach that uses long-read whole-genome sequencing data. In this method, the data is partitioned into two haplotypes using ultralong nanopore sequencing reads and barcoded short reads from long DNA molecules. Specifically, the team used WhatsHap to combine long-range phasing information from these reads, and then separated the circular consensus long reads into haplotypes for the diploid assembly.
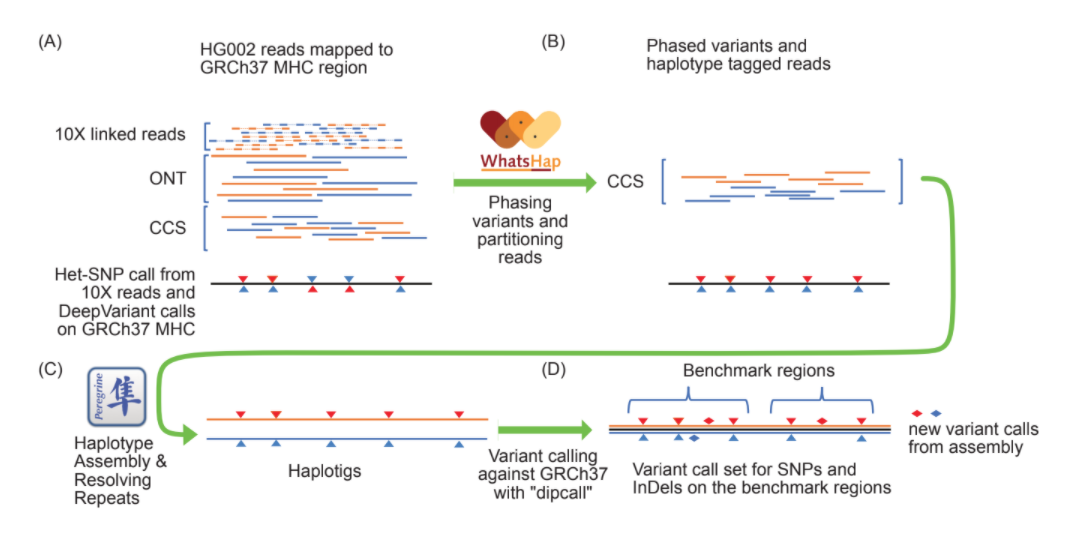
The next step was to “assemble both haplotypes of the MHC and use dipcall to generate benchmark variants and regions” in the GIAB sample, the researchers wrote. Specifically, they used dipcall to call variants by aligning the main haplotigs to the GRCh37 reference and using defined benchmark regions that excluded structural variants, extremely divergent regions, low quality regions, and long homopolymers.
The final benchmark set consists of phased small and structural variants. They formed small variant benchmark regions covering 94 percent of the MHC, with 49 percent – over 7,300 – more variants than was possible with previous mapping-based benchmarks. The benchmark regions include 22,368 benchmark SNVs and indels smaller than 50 bp, and covers almost all 23 HLA genes with some exceptions due to the high degree of variability in specific portions of genes.
To check the accuracy of the benchmark set, the team compared their assembly-based diploid variant calls to 11 variant call sets generated using various methods and sequencing technologies. The results of the comparison revealed the benchmark reliably identified false positives and false negatives across methods and technologies. In addition, they found “high concordance” between the results of their assembly-based benchmark and existing mapping-based benchmarks within regions accessible to mappers. The additional variants that the assembly-based method found “are likely from those regions where HG002 has at least one haplotype that is highly diverged from the reference, making it challenging to map individual reads.”
“This benchmark was critical for the precisionFDA Truth Challenge V2 we held this summer, which highlighted performance of variant callers in the MHC,”
Justin Zook, Human Genetics Team Lead at NIST.
These are promising results for efforts to understand and map other highly variable parts of the genome. But there is still plenty of room for improvement. “Since most of the MHC alternate loci in GRCh38 and other MHC sequences are not fully continuous assemblies, our assembled haplotigs represent two of only a few continuously assembled MHC haplotypes,” the researchers wrote. “We expect this curated benchmark set from a targeted diploid assembly will help the community improve variant calling methods and whole genome de novo assembly methods and form a basis for future diploid assembly-based benchmarks.”
Furthermore, with initiatives such as the Human Pangenome Reference Consortium which aims to sequence 350 human genomes using long read technology, “our knowledge about the whole MHC region will increase rapidly,” the researchers wrote. “A pan-genomics variant call benchmark for many individuals may become essential for economically genotyping the whole MHC region correctly.”
Meanwhile, there are other highly variable regions that would benefit from a benchmark set including the KIR and IGH loci as well as segmental duplications. Moving forward, “a combination of long read and short read technologies for resolving difficult genomic regions in many individuals will become important,” according to the team. “We hope the rich collections of diverse data sets and analyses for the GIAB samples and the future population-scale de novo sequencing will enable precision medicine from complicated genomic regions like MHC.” You can read the full paper here.


.png)
.png)
.png)