

The DNAnexus science internship has sought to mentor students, from undergraduates to Ph.D. candidates, in bioinformatics since 2015. This project was performed by Adeline Petersen during her internship with DNAnexus under the supervision of Samantha Zarate.
We are currently recruiting summer 2019 interns. If you are interested in interning with the DNAnexus science team, please apply here.
RNA-sequencing, or RNA-seq, is an important tool used to characterize and analyze the RNA transcripts present in a sample. Utilizing next-generation sequencing (NGS) technology, RNA-seq boasts advantages over previous transcriptome profiling technologies because it has higher sensitivity and precision in measuring gene expression than other hybridization-based approaches. Given the decreasing cost of NGS, the popularity of RNA-seq is on the rise: the publications on PubMed NCBI mentioning RNA-seq have increased from only 6 articles in 2008 to 3580 articles published in 2017, and the targeted RNA-seq market has been listed as a major growth area up to 2024. The clinical applications of RNA-seq range from detection of mutations in tumor and other inherited diseases to comparative gene expression analysis. Accordingly, interest in the questions that RNA-seq workflows can answer has increased among our customers. Here we present to the community an RNA-seq workflow that we developed to work out-of-the-box, as well as benchmarks demonstrating our workflow’s speed and accuracy.
Introduction
Given the rise in popularity of RNA-seq, many research institutions have published best-practices RNA-seq pipelines. The ExaScience Life Lab in Leuven, Belgium has published Halvade-RNA, a parallel, multi-node pipeline that primarily focuses on variant calling based on the GATK best practices; the Broad Institute has also published an RNA-seq best-practices short-variant calling workflow. These variant calling workflows allow users to focus on identifying variants in genes expressed, as RNA editing can lead to variation between the genes encoded in DNA and those expressed in RNA. ENCODE has similarly published different RNA-seq read quantification pipelines, all of which leverage STAR for read alignment and RSEM for read quantification. Measuring read quantification can help researchers establish a healthy baseline for gene expression and therefore identify disease progression over time. Both variant calling and read quantification are common uses of RNA-seq data, so we wanted to implement both tasks in our workflow.
The QuickRNASeq model, developed by Pfizer, focuses on both variant calling and read quantification using large-scale RNA-seq data. QuickRNASeq generates accessible data analyses and visualizations by using STAR for read alignment, featureCounts from the Subread package for read quantification, VarScan for germline variant calling, and RSeQC for quality control of RNA-seq data. We chose to model our first attempt at a DNAnexus-recommended RNA-seq workflow after the QuickRNASeq model because of its versatility, accuracy, runtime, and emphasis on large-scale data input.
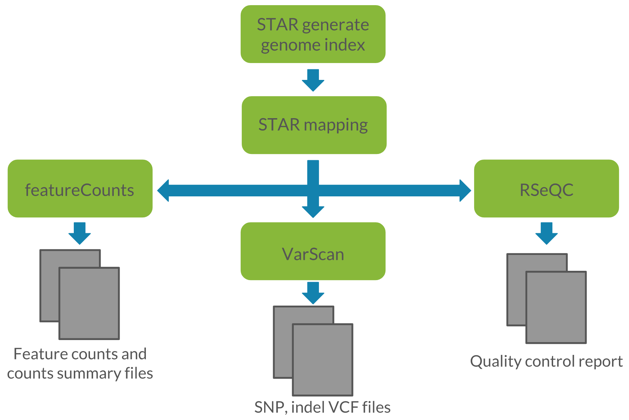
Figure 1: Visualization of the proposed workflow. We utilized STAR for read alignment, featureCounts for read quantification, VarScan for germline variant calling, and RSeQC for quality control of RNA-seq data.
Features of the Workflow
STAR
STAR, or Spliced Transcripts Alignment to a Reference, is used to map reads to a reference genome. In particular, STAR lends itself well to RNA-seq analysis by accepting an annotations file in GTF format, allowing it to detect splice junctions within a read.
We compared STAR v2.6.1a, published as of August 2018, with v2.5.3a and found that these metrics were nearly identical for both the generate genome index and mapping steps (Figure 2).
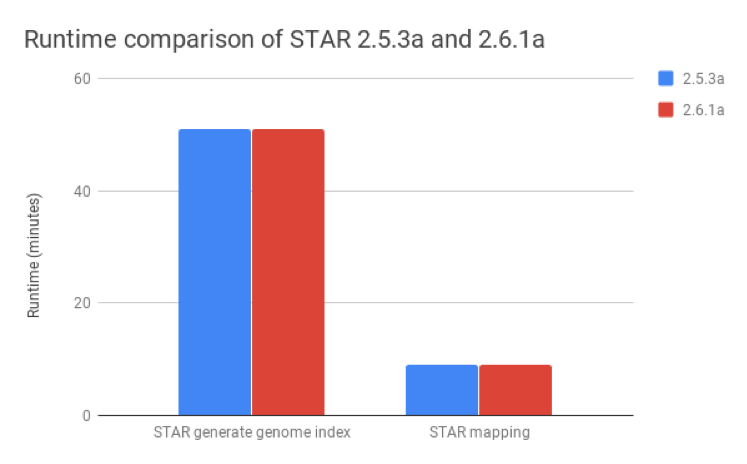
Figure 2: Runtime comparison between STAR versions 2.5.3a and 2.6.1a. The runtimes for the generate genome index and mapping steps are nearly identical for both versions.
featureCounts
We chose featureCounts v1.6.2 from the Subread package as the application for read quantification. This application is an efficient and general-purpose read summarization program, and counts mapped reads to specific genomic features. Furthermore, featureCounts is both accurate and specific, accounting for insertion/deletions, junctions, and structural variants. As input, featureCounts uses the alignment file from STAR (the genome BAM file) and an annotation file. It outputs a read counts file and a read counts summary file.
VarScan
We chose VarScan v2.4.3 as the germline variant calling application. VarScan is a platform-independent variant calling program that utilizes samtools mpileup. It can call germline variants, somatic variants, copy number variants, and de novo mutations. For our application, we used the germline variant calling functionality of VarScan to identify SNPs and insertions/deletions present in the RNA-seq input data compared to the reference genome. This application takes the genome BAM from STAR, a reference genome, and an annotation file as inputs; its primary output is a VCF file containing SNPs and indels.
RSeQC
We chose RSeQC v2.6.5 as the quality control application for the workflow. Quality control is particularly important for RNA-seq analyses because the transcripts present in a sample can vary drastically over time, between cell types in a sample, and between physiological conditions of a sample. Therefore, verifying the quality of the RNA-seq data through reproducibility, as is typically done with DNA sequencing, is challenging. RSeQC includes a number of metrics to ensure the quality of alignment, sequencing, and library preparation. It also assesses the feasibility of performing alternative splicing analysis. RSeQC takes the genome BAM file from STAR as an input, along with a 12-column BED file as the annotation file. RSeQC outputs a PDF file containing metrics and plots for analyzing alignment and sequencing quality, a sampling of which is shown in Figure 3.

Figure 3: Example sample graphics from the RSeQC quality control report.
3a: clipping profile showing clipped read alignment.
3b: gene body coverage plot showing high average coverage over the transcriptome.
3c: heat map showing nucleotide Phred quality score over read, where blue represents low nucleotide density and red represents high nucleotide density.
3d: junction saturation plot, showing medium RNA-seq data saturation.
Benchmarking
At present, there does not yet exist a “truth set” of read quantities we can use as a benchmark. Therefore, because ENCODE has been leading the effort towards uniform processing pipelines, we chose to compare our workflow against it for our benchmarking. We benchmarked our workflow against the ENCODE RNA-seq workflow, which had also been implemented on DNAnexus and used a different version of STAR for alignment and RSEM for read quantification. We modified our own workflow (Figure 1) to provide a more even comparison, paring it down to utilize STAR for alignment and featureCounts for read quantification.
We found that the read quantification results correlated closely between our workflow and the ENCODE workflow, with a mean correlation coefficient of 0.895 and minimum 60,000 reads mapped (Figure 6). The workflow we developed was nearly twice as fast as the ENCODE workflow, with much of the benefit coming from our implementation of featureCounts (Figure 7). As a result, we can be confident of both the results and efficiency of our workflow.
However, we must note that RSEM and featureCounts are not identical programs: RSEM performs transcript and read quantification, whereas featureCounts performs read quantification only, meaning that RSEM can give users more information than what we used to perform this read quantification analysis. This may explain the differences in runtime between our workflow and that of ENCODE.
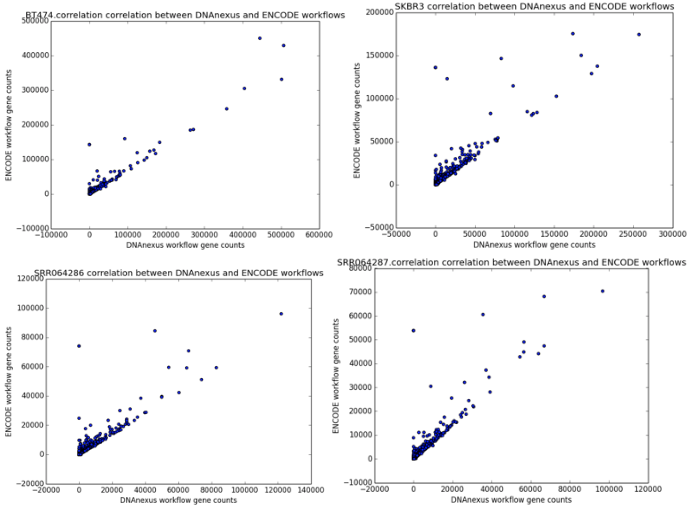
Figure 4: Correlation scatter plots between read counts generated by our workflow (x-axis) and read counts generated by the ENCODE workflow (y-axis). The four scatter plots represent the outputs from the samples BT474, SKBR3, SRR064286, and SRR064287.
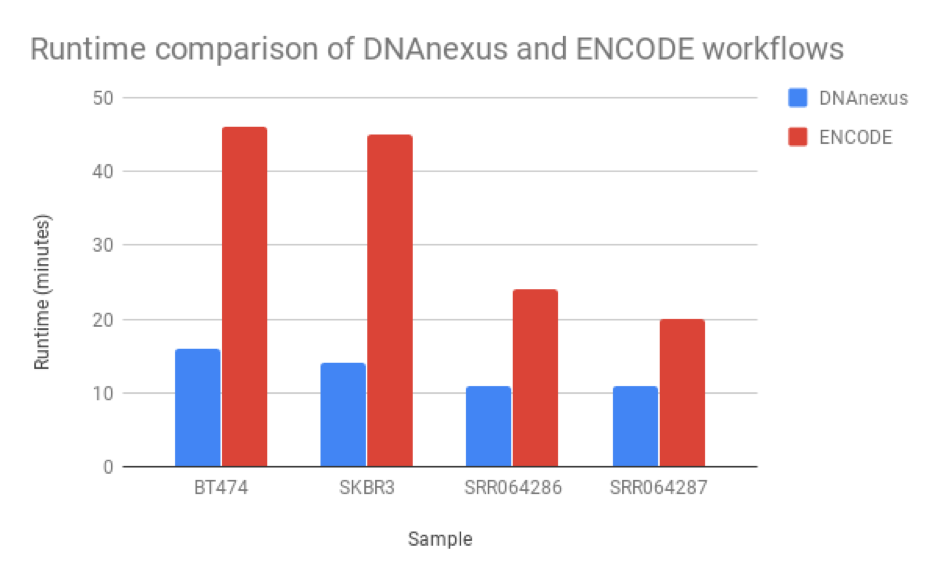
Figure 5: Runtime comparison between the workflow we developed and the ENCODE workflow. Our workflow runs at least 2X faster than the ENCODE workflow, though this may be due to RSEM’s ability to perform isoform as well as read quantification.
Final Notes and Conclusion
DNAnexus has worked with many customers on RNA-seq workflows. Through this experience, we decided that developing a workflow to make publicly available and to universally recommend to customers would benefit the scientific community as a whole. We are committed to providing users with the most up-to-date tools and standards available, and this preliminary effort represents a foray into providing best-practices workflows accordingly.
We generated the data presented in these benchmarks and figures using the SRA database (SRR064286 and SRR064287) as well as BT474 and SKBR3 samples.
The workflow is currently available upon request; please contact support@dnanexus.com to obtain access. In the future, we plan on releasing or updating all applications mentioned here on the DNAnexus app library and creating a Featured Project on the platform showcasing the workflow and giving users sample data on which they can run this workflow. We also plan on investigating other RNA-seq analysis tools such as Sailfish, Kallisto, and Salmon in the future.
This research was performed by Adeline Petersen as part of her internship with DNAnexus. The project was supervised by Samantha Zarate and assisted by Yih-Chii Hwang, Ph.D., Alpha Diallo, Ph.D., Naina Thangaraj, Steve Osazuwa, Brett Hannigan, Ph.D., and Andrew Carroll, Ph.D.


.png)
.png)
.png)