
The detection of somatic mutations in sequenced cancer samples has become increasingly standard in research and clinical settings, as they provide insights into genomic regions which can be targeted by precision medicine therapies. Due to the heterogeneity of tumors, somatic variant calling is challenging, especially for variants at low allele frequencies. Researchers use common somatic variant call tools, including MuTect, MuSE, Strelka, and Somatic Sniper, that detect somatic mutations by conducting paired comparisons between sequenced normal and tumorous tissue samples. Each of these variant callers differ in algorithms, filtering strategies, recommendations, and output. Thus we set out to compare how these individual apps perform on the DNAnexus Platform. Each app was evaluated for recall and precision, cost, and time to complete.
To benchmark some of the common somatic variant calling tools available on the DNAnexus Platform, our team of scientists simulated synthetic cancer datasets at varying sequencing depths. DNA samples from the European Nucleotide Archive were obtained and mapped to the hs37d5 reference with the BWA-mem FASTQ read mapper on DNAnexus.
These samples were then merged into a single BAM file representing the normal sample. To obtain the tumor sample, synthetic variants were inserted into each individual sample with the BAMSurgeon app on DNAnexus. All simulated samples were then merged into one BAM file constituting the tumor sample. Both the synthetic tumor and normal BAM files had approximately 250X sequencing depth.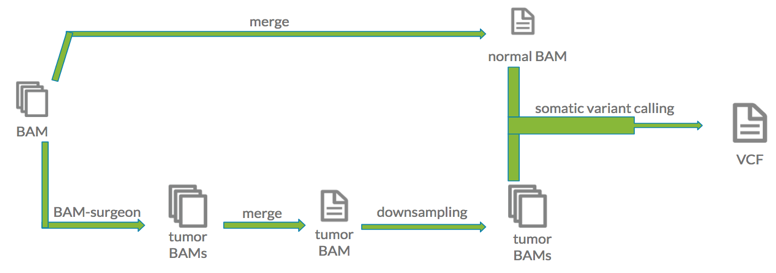 The synthetic tumor BAM file was then downsampled into a range of sequencing depths. With the help of sambamba through the Swiss Army Knife application, these files were reduced to 5X, 10X, 15X, 20X, 30X, 40X, 50X, 60X, 90X, and 120X coverage files. The file representing the normal sample was downsampled into a 30X sequencing depth file. Once the synthetic cancer dataset was created, the common somatic variant calling tools MuTect, MuSE, Strelka, and Somatic Sniper were run to detect single nucleotide variants. Upon completion, the high quality variants were filtered from each VCF.
The synthetic tumor BAM file was then downsampled into a range of sequencing depths. With the help of sambamba through the Swiss Army Knife application, these files were reduced to 5X, 10X, 15X, 20X, 30X, 40X, 50X, 60X, 90X, and 120X coverage files. The file representing the normal sample was downsampled into a 30X sequencing depth file. Once the synthetic cancer dataset was created, the common somatic variant calling tools MuTect, MuSE, Strelka, and Somatic Sniper were run to detect single nucleotide variants. Upon completion, the high quality variants were filtered from each VCF.
Results:
Recall
MuTect performed the best at classifying correct variants followed by Strelka, MuSE, and Somatic Sniper. This was consistent across allele frequency thresholds of 01, 0.2, 0.3, 0.4, and 0.5.
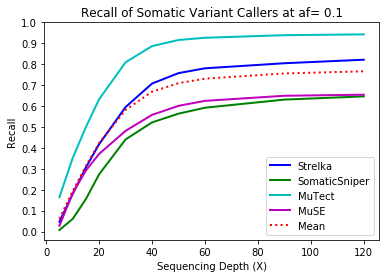
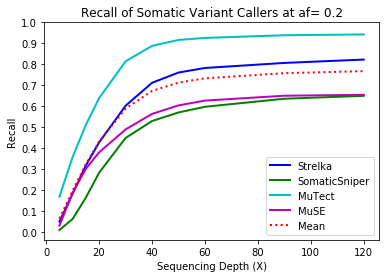
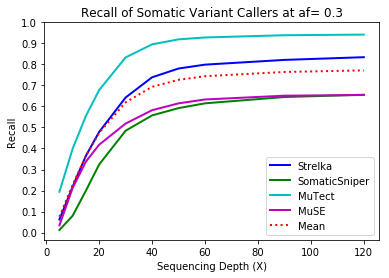
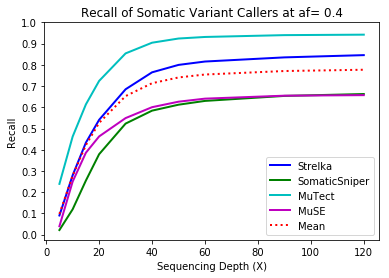
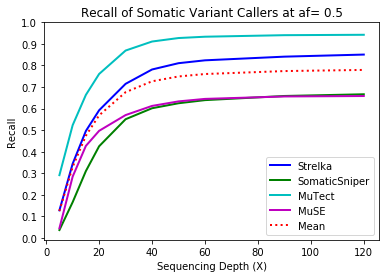
Coverage and Recall
One interesting finding – for the callers investigated, the ability to recall variants at lower frequencies showed a similar pattern. Each of the callers discovers more of the variants before plateauing at a recall ceiling at a certain coverage. Lower allele frequencies require more coverage before saturating for recall at a caller. 30-fold coverage was required to reach the plateau of 0.5 allele frequency variants, while 40-fold coverage was required for 0.1 allele frequency variants. Reliable detection of lower frequency variants presumably require still more coverage to reach a recall plateu.
Precision
All tools performed well at identifying relevant variants (>95% precision) regardless of tumor sequencing depth.
To get a more accurate view of the interplay between precision and recall, the harmonic mean of precision and recall (F-score) was computed for each output VCF by depth. MuTect had the best performance overall, followed by Strelka, and then MuSE, and Somatic Sniper.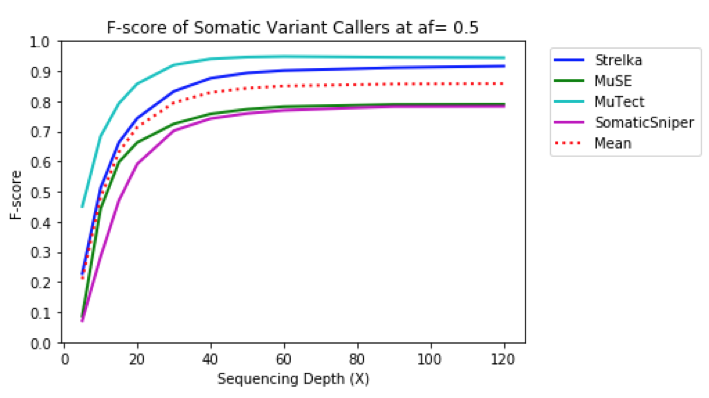 Runtime & Cost
Runtime & Cost
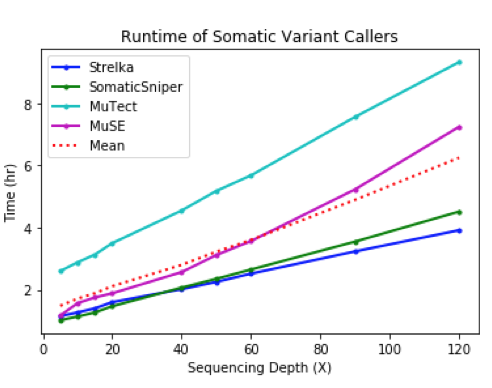
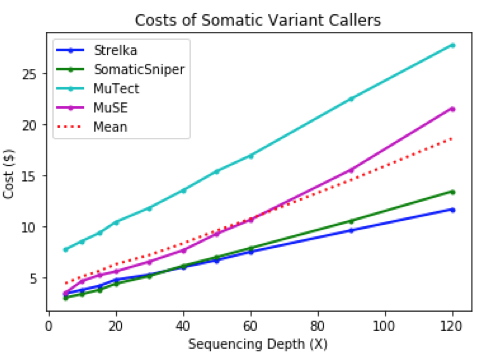
Out of all the apps, Strelka finished most rapidly for the lowest cost. Compared to MuTect, Strelka did not score as high for precision or recall, but completed the analysis of single nucleotide variants in a fraction of the time.
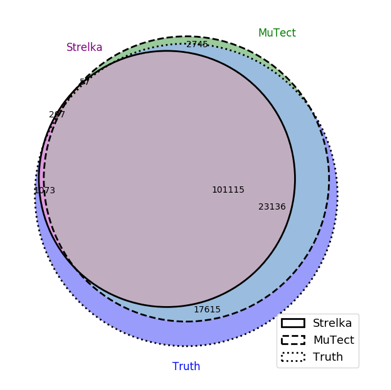
To get a more detailed comparison between MuTect and Strelka, this 3-way venn diagram compares these tools to the truth set. Note, the false negatives called by MuTect are likely due to noise in the dataset.
 To better visualize the differences between the callers, we converted the output of each of the callers into high-dimensional vectors in which each variant call in any of the samples is one of the dimensions. This format allows us to calculate the distances between each of the programs and with the truth set. This also allows us to use standard methods such as Mulitdimensional Scaling to convert these distances into positions in 2-D space (axes units are arbitrary, only relative position matter is the graph below).
To better visualize the differences between the callers, we converted the output of each of the callers into high-dimensional vectors in which each variant call in any of the samples is one of the dimensions. This format allows us to calculate the distances between each of the programs and with the truth set. This also allows us to use standard methods such as Mulitdimensional Scaling to convert these distances into positions in 2-D space (axes units are arbitrary, only relative position matter is the graph below).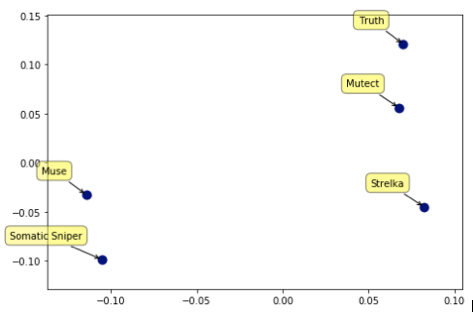
Valid variant calling results are crucial as next-generation sequencing data is increasingly applied to the development of targeted cancer therapeutics. Our analysis of MuTect, MuSe, Strelka, and Somatic Sniper found that the best results with respect to precision and recall can be achieved by using MuTect. Strelka was also a top performer, and simultaneously reduced runtime and cost.
Need to detect variants in your dataset? Get started using these tools on DNAnexus today.
This research was performed by Nicholas Hill and Victoria Wang as part of their internship with DNAnexus. The project was supervised by Naina Thangaraj, Arkarachai Fungtammasan, Yih-Chii Hwang, Steve Osazuwa, and Andrew Carroll.


.png)
.png)
.png)
{kind=link}