


In this blog we discuss the newly published use of PacBio Circular Consensus Sequencing (CCS) at human genome scale. We demonstrate that DeepVariant trained for this data type achieves similar accuracy to available Illumina genomes, and is the only method to achieve competitive accuracy in Indel calling. Early access to this model is available now by request, and we expect general availability in our next DeepVariant release (v0.8).
Editorial Note: This blog is published with identical content here and on the Google DeepVariant blog. Re-training of DeepVariant and accuracy analyses were performed by Alexey Kolesnikov, Pi-Chuan Chang, and Andrew Carroll from Google. Sequence context error analysis was performed by Jason Chin of DNAnexus.
PacBio Circular Consensus and Illumina Sequencing by Synthesis
The power of a sequencing technology (e.g. accuracy, throughput, and read length) is determined by its underlying biochemistry and physical measurement. Each read in Illumina sequencing by synthesis (SBS) corresponds to clustered copies of the same DNA molecule. The consensus of an SBS cluster provides high accuracy, but molecules in the cluster go out of phase longer in the read, ultimately limiting read lengths.
The ability of PacBio’s single molecule real-time (SMRT) sequencing to measure a single DNA molecule allows it to escape this limitation on read length (important in applications like genome assembly, structural variation, and in difficult regions). However, sampling a single molecule without a consensus is more error prone, with base error rates of 10-15%.
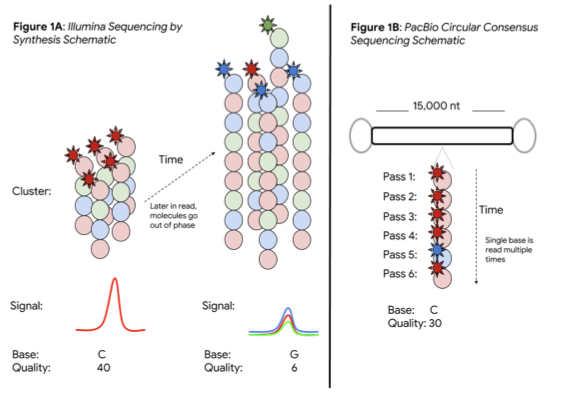 PacBio CCS builds a consensus on the same base. A stretch of DNA with a controlled length (e.g. 15,000 bases) is linked by known adapters. Sequencing DNA multiple times provides the best of both worlds: a long read length and a measurement, reaching 99% per-base in a consensus read. This promises a single data type strong for both small variant analysis and structural variation.
PacBio CCS builds a consensus on the same base. A stretch of DNA with a controlled length (e.g. 15,000 bases) is linked by known adapters. Sequencing DNA multiple times provides the best of both worlds: a long read length and a measurement, reaching 99% per-base in a consensus read. This promises a single data type strong for both small variant analysis and structural variation.
Although the PacBio CCS base error rates are low, the sequence context of the errors differs from Illumina’s, and variant callers need modification to perform optimally. Instead of being coded by humans, DeepVariant learns which features are important from the data. This unique attribute allows it to be quickly adapted to PacBio CCS by re-training on this data.
Re-Training DeepVariant for PacBio CCS
Training DeepVariant involves starting from a model checkpoint and showing it labeled examples. This changes the weights of the model over time. We started from a DeepVariant Illumina WGS model and re-trained this with PacBio CCS data, excluding chromosome 20 to allow this to be used for independent evaluation. The PacBio CCS reads were generated from HG002, which has a truth set available from Genome in a Bottle and is the basis for training. (To learn more, see this walkthrough on training).
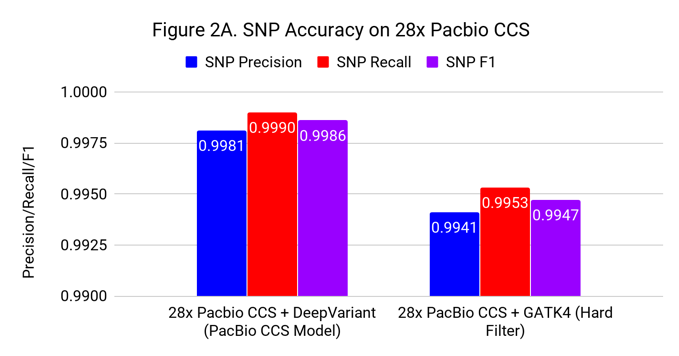 Figures 2A and 2B show the SNP and Indel performance on PacBio CCS for DeepVariant after retraining for CCS and GATK4 run with flags and filters chosen by PacBio to improve performance for CCS.
Figures 2A and 2B show the SNP and Indel performance on PacBio CCS for DeepVariant after retraining for CCS and GATK4 run with flags and filters chosen by PacBio to improve performance for CCS.
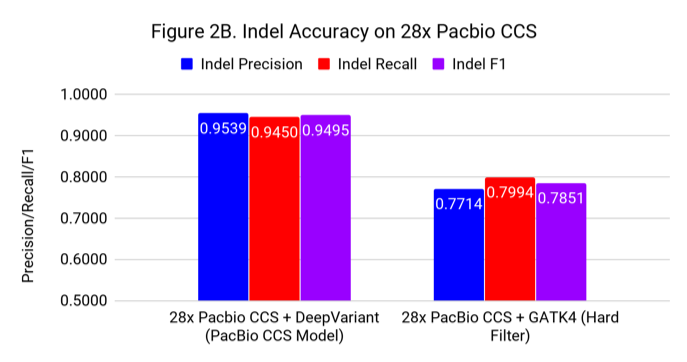 The difference for SNP calling between GATK4 and DeepVariant is similar to what we see with Illumina. However, the gap in indel performance is substantial, highlighting the need to adapt existing methods. (Note the use of different y-axes) .
The difference for SNP calling between GATK4 and DeepVariant is similar to what we see with Illumina. However, the gap in indel performance is substantial, highlighting the need to adapt existing methods. (Note the use of different y-axes) .
To put these accuracies into context, we compare to SNP and Indel F1 scores for 30x Illumina genomes from PCR-Positive and PCR-Free preparations. The PCR-Positive is the NovaSeq S1: TruSeq 350 Nano sample available on BaseSpace, evaluated on chr20. The PCR-Free is a 30x downsample of our WGS case study. Figure 3 places the accuracy of PacBio CCS as roughly in-between Illumina PCR-Free and PCR-Positive.
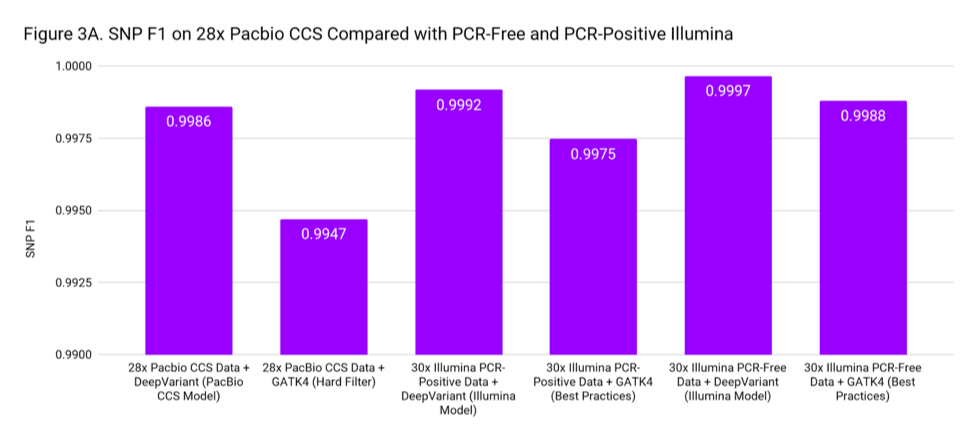
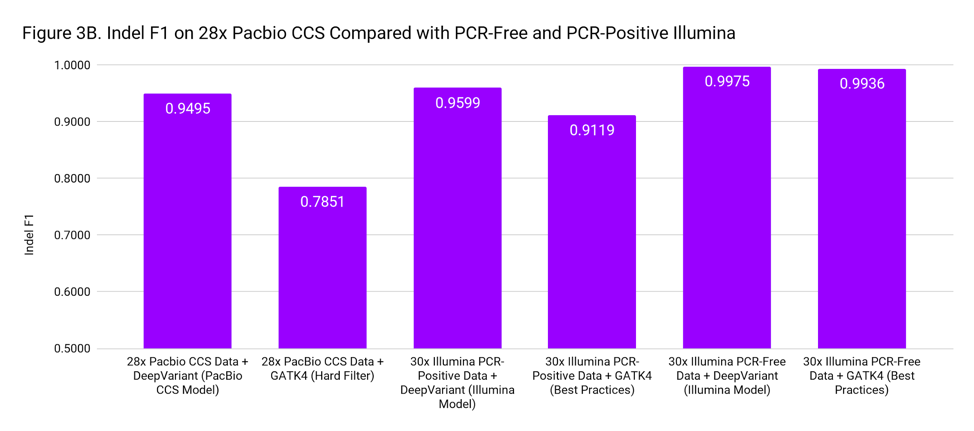 These accuracies are based on the Genome in a Bottle confident regions. The superior mappability of the PacBio CCS reads likely means this accuracy can be achieved over more of the genome (including clinically important genes) than with short reads.
These accuracies are based on the Genome in a Bottle confident regions. The superior mappability of the PacBio CCS reads likely means this accuracy can be achieved over more of the genome (including clinically important genes) than with short reads.
How Much PacBio CCS Coverage is Necessary
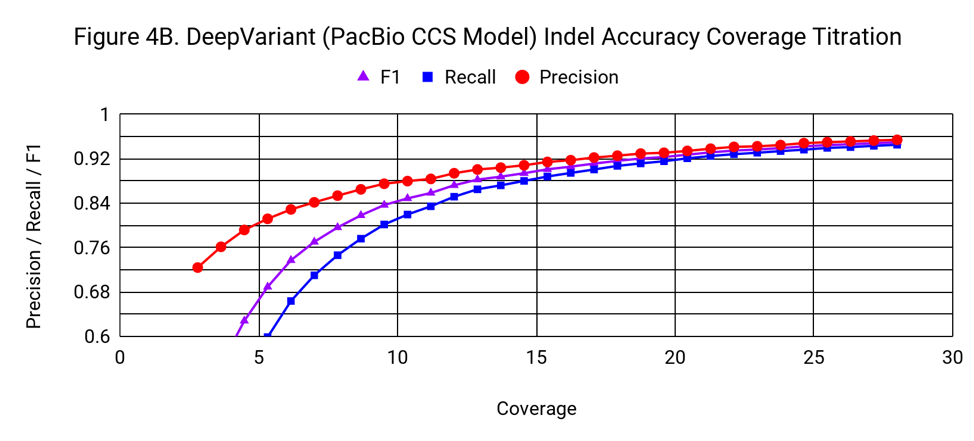
To understand how the accuracy of DeepVariant relates to coverage, we progressively downsampled from the 28x starting coverage, randomly using 3% fewer reads with each step.
SNP accuracy is quite robust to downsampling, down to a coverage of around 15x. DeepVariant’s SNP F1 at 13.7x coverage is 0.9957, exceeding GATK4’s F1 at 28x (0.9951).
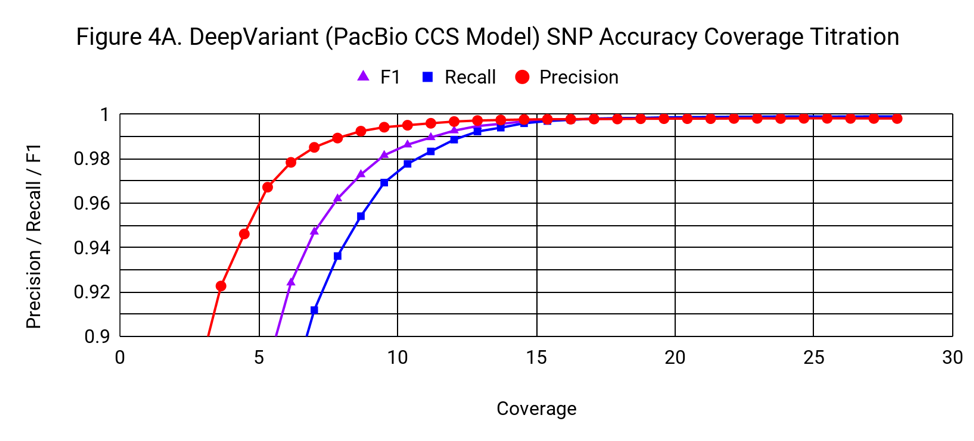
Indel accuracy declines with a gradual, but noticeable slope as coverage drops from 28x. It crosses the threshold of 0.9 F1 at about 15x.
Improving Calls by Adding Phased Haplotype Information
The ability to determine whether two nearby variants are present on the same DNA molecule (e.g. both on the copy inherited from the mother) or on different molecules is called phasing. Longer read lengths improve the ability to phase variants, as tools like WhatsHap demonstrate for PacBio reads.
PacBio uploaded CCS reads annotated with phase information using inheritance from the trio and 10X data. We incorporated this information by sorting the reads in the pileup based on their haplotype, which reorders all of the tensors (e.g. base, strand, MAPQ).
This might sound like a small change, but it may have a substantial impact on how information flows through DeepVariant’s Convolutional Neural Network (CNN). The lowest layers of a CNN see local information. Sorting reads by haplotype means that even at the lowest level, the network can learn that adjacent reads likely come from the same haplotype.
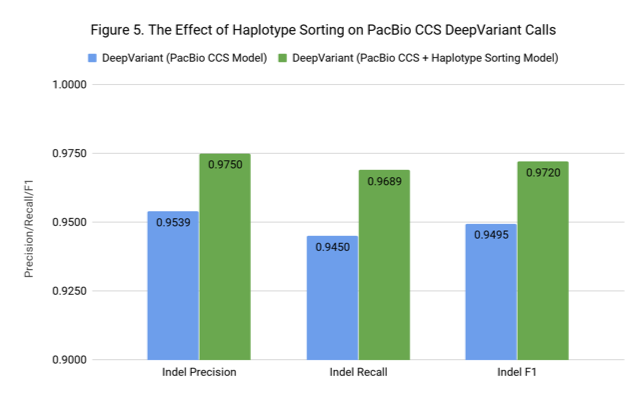 Haplotype sorting had a small positive impact on SNPs, improving F1 from 0.9986 to 0.9988. However, as Figure 4 shows, the effect for Indel F1 was large, increasing F1 from 0.9495 to 0.9720. Now that we know haplotype information has such a strong positive effect, we can consider how to add this using only the PacBio data.
Haplotype sorting had a small positive impact on SNPs, improving F1 from 0.9986 to 0.9988. However, as Figure 4 shows, the effect for Indel F1 was large, increasing F1 from 0.9495 to 0.9720. Now that we know haplotype information has such a strong positive effect, we can consider how to add this using only the PacBio data.
Using Nucleus for Error Analysis
Jason Chin of DNAnexus (and formerly of PacBio) performed several interesting analyses on top of the open-source Nucleus developed to simplify bringing genomics data into TensorFlow. See this Jupyter Notebook for a hands-on demonstration using data from a public DNAnexus project to fetch the sequence context around false positive and false negative sites.
We encoded the sequence alignment of the flanking regions (totaling 65 bases) of each site as two 65 x 4 matrices. The matched, mismatched or missing bases in the alignments were encoded to the first 65 x 4 matrix. In this matrix, each column had 4 elements and counts the number of A/C/G and T bases of the reads that match the reference at a given location. The inserted sequences relative to the reference were encoded into the second 65 x 4 matrix.
We collected the error context matrices and treated them like high dimension vectors. We applied common dimensionality reduction techniques to see if we could find common patterns around the erroneous sites. Indeed, intriguing clustering structures appeared when we applied T-SNE or UMAP (see the figure below). While long homopolymer A or T sequence cause the majority of the errors, we observed other less trivial common patterns, e.g., di-nucleotide or tri-nucleotide repeats. We also discovered a set of reads that have approximately a common prefix that is corresponding to Alu repeats.
Figure 6. A number of repeats patterns identified by a UMAP embedding of the alignment vectors around the residue error sites.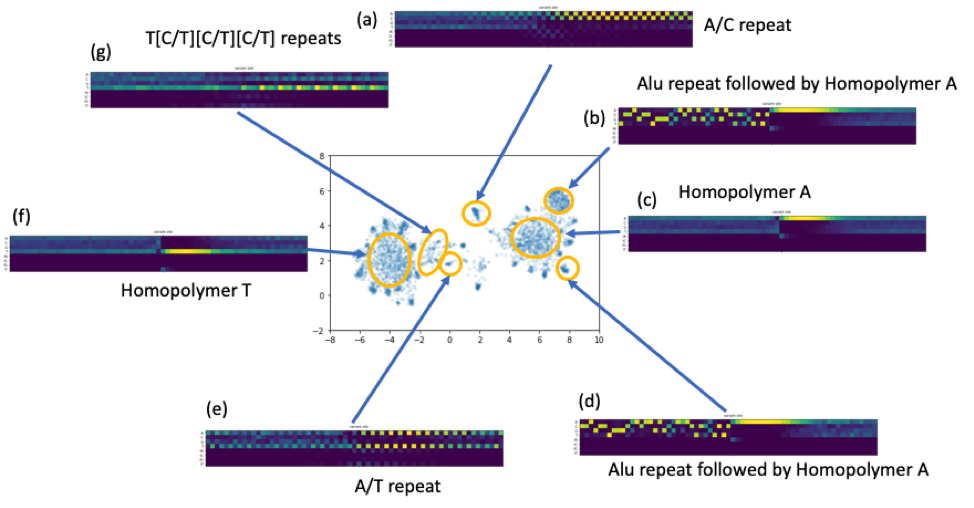
Future Work
The models generated for this analysis are currently available by request to those considering CCS for their workflows. We expect to make a PacBio CCS model fully available and supported alongside our Illumina WGS and exome models in the next DeepVariant release (v0.8).
Though we feel the current work is strongly compelling for use, we identified a number of areas for continued improvement. Currently, we have trained with examples from only one CCS genome on a single instrument, compared to 18 Illumina genomes from HiSeq2500, HiSeqX, and NovaSeq. Simply having more training examples should improve accuracy.
The ability to generate phasing information solely from the PacBio reads would provide another large gain. We are investigating whether similar approaches are possible to improve DeepVariant for Illumina data in variant-dense regions. Finally, hybrid Illumina-PacBio models are an intriguing possibility to explore.
We have continued to improve DeepVariant’s speed and accuracy, and we expect to achieve similar improvements on PacBio CCS data as it becomes widely used.
Requesting Early Access
To request early access to the PacBio CCS model generated for this work, you can email awcarroll@google.com. We expect general availability of this model alongside our Illumina WGS and Illumina exome models in our next DeepVariant release (v0.8).
The app is also available by request on DNAnexus. For access please email support@dnanexus.com. This app will be broadly available soon.
A Consensus of Scientific Expertise
The CCS manuscript investigates other applications: structural variant calling, genome assembly, phasing, as well as the small variant calling discussed here. This required bringing together many investigators with different specializations across both the wet lab and informatics.
All of these investigators play important roles in evolving PacBio CCS into wide application. We want to specially thank Billy Rowell from PacBio, who coordinated the small variant calling section, and Aaron Wenger, who coordinated the broader manuscript, and Paul Peluso and David Rank, who generated the CCS dataset. We also give special thanks to Jason Chin, who helped to bring our team into this investigation, and who has been responsible for advancing many cutting edge PacBio applications over the years.




