
The application of Deep Learning methods has created dramatically stronger solutions in many fields, including genomics (as a recent review from the Greene Lab details). In this blog, we focus on a different aspect – the ability of deep learning to empower those with domain insight to rapidly create methods for new technologies or problems.
Last weekend, two teams with members from DNAnexus participated in a SV.AI hackathon chaired by Ben Busby, who leads a series of NIH Hackathons. NCI and GDC also helped a great deal.
For this hackathon, Bill Paseman, a patient with renal cell carcinoma and the founder of RareKidneyCancer.org for advancing rare kidney cancer research and patient advocacy, donated both sequence data from tumor cells and the corresponding whole blood sequence. This sequence data was >90x coverage from the BGI-SEQ 500 instrument.
The hackathon itself was stellar, and all presentations and data are available on GitHub including those from both DNAnexus teams, CNN and RNN. Another group was able to provide a very comprehensive analysis of the tumorigenesis.
Because the BGI-SEQ is such a new instrument, and because public deep coverage WGS from it is difficult to find, the RNN team decided to use the hackathon to evaluate the analytical methods being used on BGI-SEQ and to explore the possibility to improve tools for this data type.
This work proved to strongly validate the concepts of deep learning. In only a single weekend, we achieved significant improvements to two separate deep learning methods.
Evaluation of BGI-SEQ
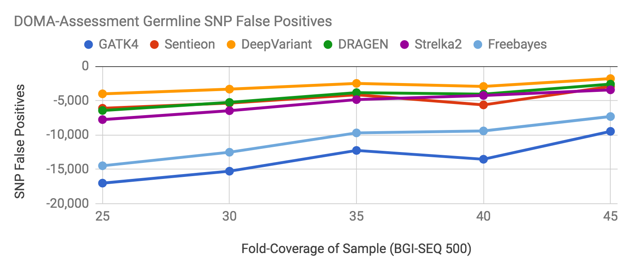 First, we attempted to assess what issues may exist in applying tools developed for Illumina technology to the BGI-SEQ 500. To do this on a genome which did not have a truth set, we developed a method called DOMA (Drop Out of Mutual Agreement).
First, we attempted to assess what issues may exist in applying tools developed for Illumina technology to the BGI-SEQ 500. To do this on a genome which did not have a truth set, we developed a method called DOMA (Drop Out of Mutual Agreement).
A more detailed description is in our presentation. We plan to release a further blog post on this method later. Values closer to 0 (higher on the chart) reflect better performance.
 As a high-level summary, we found error rates were about 3x higher in SNP calling and 10x higher for indel calling. False negatives for indel calling was the dominant error mode, consistent with the only 30x BGI-SEQ Genome in a Bottle sample.
As a high-level summary, we found error rates were about 3x higher in SNP calling and 10x higher for indel calling. False negatives for indel calling was the dominant error mode, consistent with the only 30x BGI-SEQ Genome in a Bottle sample.
Some methods performed relatively worse in this evaluation. For example, it is unusual to see GATK depart so greatly from Sentieon.
Minding the Gap: Why Performances Differ
The implication here is that components of the BGI-SEQ technology are different enough from Illumina that the methods built with Illumina in mind either can’t take advantage of new information in BGI-SEQ, don’t know how to mitigate BGI-SEQ weaknesses, or bring biases that don’t apply to BGI-SEQ data. Even a quick visual inspection hints at this:
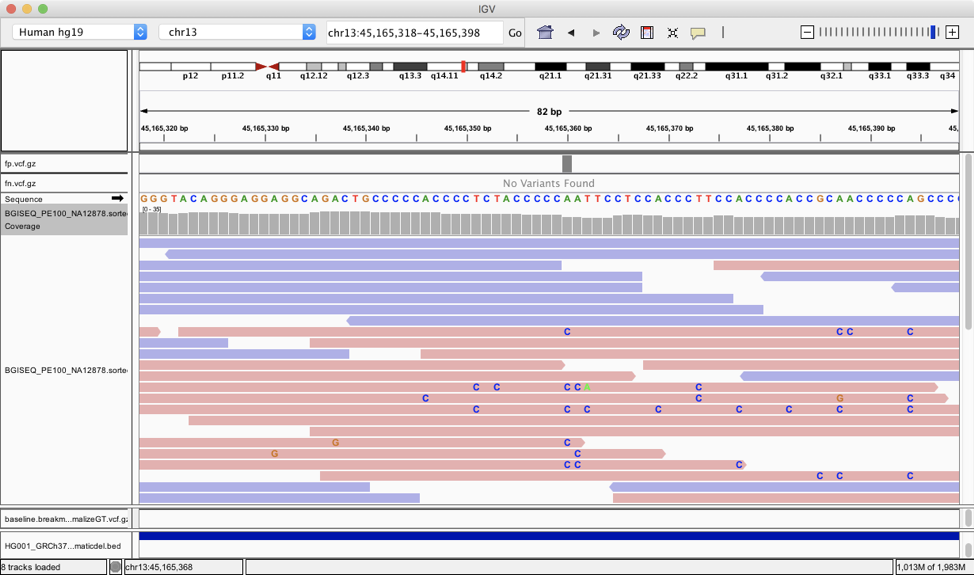
In this GC rich region, it appears there are some systematic A/T > C errors that only happen in those reads sequenced from 5’ to 3’. While this may not be prevalent, it could cause false positive variant calls especially the sequence coverage is low.
There are currently two published variant calling tools that use deep learning methods: DeepVariant and Clairvoyante. We were able to train both of them on BGI-SEQ data.
Training a BGI-SEQ model for Clairvoyante
Clairvoyante was published in bioRxiv just a couple weeks ago, led by Ruibang Luo, an assistant professor at the University of Hong Kong who developed Clairvoyante as a postdoc in Michael Schatz’s lab.
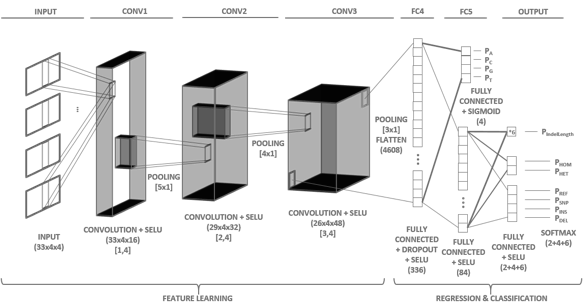 Clairvoyante uses a less complex architecture than DeepVariant, allowing it to be trained using less hardware and to make variant calls quickly. Though its accuracy on Illumina data is not as strong, the framework can call variants on long-read data. It is likely the strongest variant caller for Pacbio and Oxford Nanopore. Ruibang graciously cites Jason’s VariantNet blog post, which discusses many of the concepts for deep learning on genomic data with similar architectures.
Clairvoyante uses a less complex architecture than DeepVariant, allowing it to be trained using less hardware and to make variant calls quickly. Though its accuracy on Illumina data is not as strong, the framework can call variants on long-read data. It is likely the strongest variant caller for Pacbio and Oxford Nanopore. Ruibang graciously cites Jason’s VariantNet blog post, which discusses many of the concepts for deep learning on genomic data with similar architectures.
To extend Clairvoyante, Jason generated a workflow and applets to reproduce preparing data for training and to call variants as reproducible pipelines on DNAnexus platform.
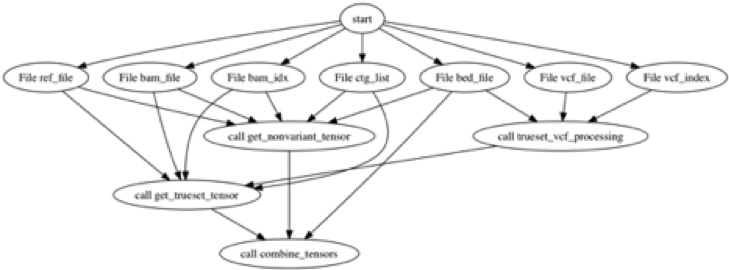
The end-to-end training to calling with Clairvoyante can be decomposed to three stages:
(1) Building training data: the workflow is configured with WDL and Docker. We use dxWDL to transform the WDL into a DNAnexus workflow for deployment. The WDL workflow (shown below) is slightly more complicated than the rest of the steps, as it involves some complicated variant file filtering steps.
(2) Model training: a native DNAnexus applet running the Clairvoyante convolutional neural network to generate trained models.
(3) Variant calling: a WDL task-based DNAnexus applet taking the trained and alignment BAM files to generate variant calls.
With this setup, we can run each step using simple dx-toolkit commands or the DNAnexus platform UI to fully reproduce the deep learning variant calling pipeline for the three technologies used as examples in the preprint and the BGI-SEQ data. Within the DNAnexus project environment, all of these can be done without explicit knowledge on how to set up the working environments for different steps. A user only has to set up the proper input files for the workflow.
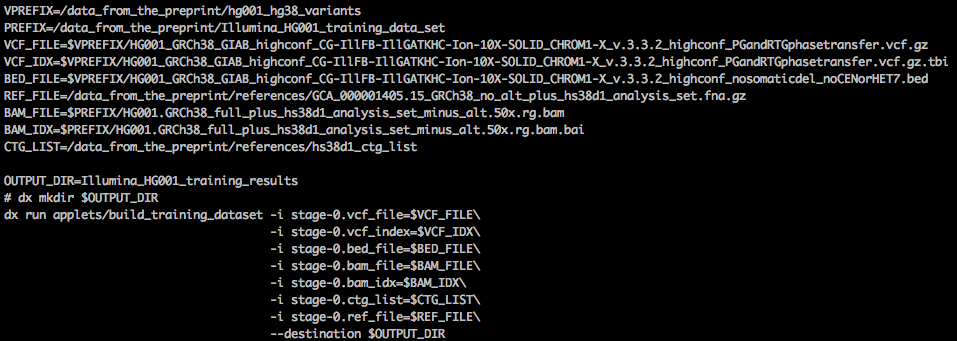
Launching the training data preparation step with the command line
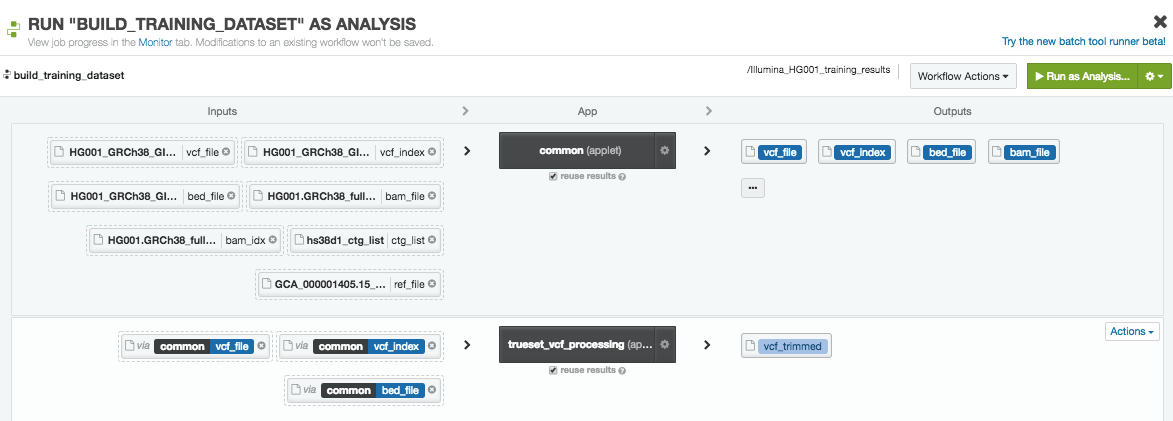
Launching the training data preparation step with the DNAnexus platform UI
All data files, trained models, variant calls, workflow and applets are available on the DNAnexus public project “clairvoyante_dnanexus_demo” to those with DNAnexus accounts. If you’d like to try running these steps yourself, you can copy the contents of the public project to a new project and execute the code in the “job_scripts” directory of the GitHub repo using the dx-toolkit command line environment.
Jason’s work on re-training Clairvoyante on DNAnexus platform is in a GitHub repo. While the code targets deployment on the DNAnexus platform, the workflow and environment setup could be adapted to a generic computation platform or cluster as well.
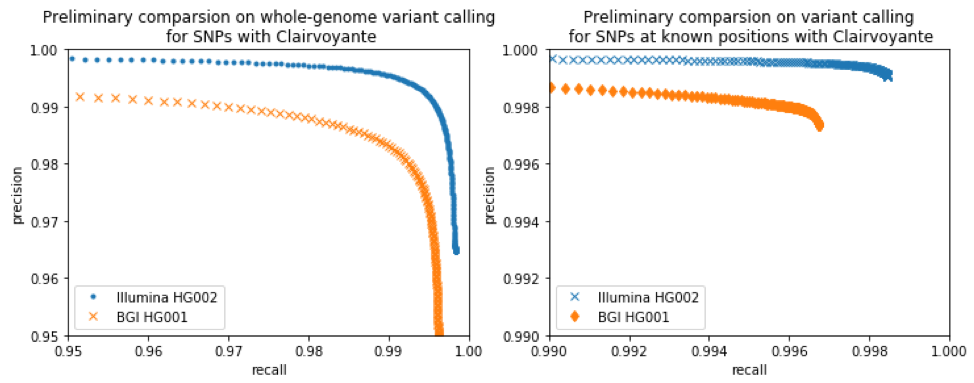
The charts below indicate the performance of Clairvoyante on Illumina and BGI-SEQ data. Clairvoyante is able to achieve a high overall accuracy on BGI-SEQ data. The slightly lower accuracy suggests that BGI-SEQ data is somewhat more challenging to use.
Thinking more broadly, genomic deep-learning model developers can use DNAnexus as a platform for continuous integration and testing. The platform provides a seamless integration platform for genomic data management and cloud job executions, which can make pipeline execution tests possible with even larger and more realistic datasets. A production center which added every validation run to a DNAnexus project could continuously build new models and create a constantly improving method tuned to the production conditions of their operation.
Training DeepVariant for BGI-SEQ
We have already discussed DeepVariant in several blogs evaluating the method, its robustness, and the Google Brain’s continued improvements to it. Pi-Chuan Chang from Google Brain demonstrated how to retrain DeepVariant.
 GoogleBrain recently released Nucleus, a framework which provides hooks from common genomic data formats into TensorFlow. Pi-Chuan’s work used only this publicly-facing resource; anyone can replicate these steps to extend DeepVariant to their data or problem of choice.
GoogleBrain recently released Nucleus, a framework which provides hooks from common genomic data formats into TensorFlow. Pi-Chuan’s work used only this publicly-facing resource; anyone can replicate these steps to extend DeepVariant to their data or problem of choice.
The document Improve DeepVariant for BGISEQ germline variant calling gives detailed, step by step instructions on this process (with screenshots).
Referencing back to Indel recall being the main error mode, the chart below shows the improvement from even a small amount of training (chromosome1 of one genome). Because this amount of data is much less than that used in the full DeepVariant training, and because we use the Illumina-trained DeepVariant as a base, we call this “fine-tuning”.
| Method | Data Type | SNP F1 | Indel F1 |
| GATK4 Best Practices | BGI-SEQ | 99.74% | 87.49% |
| DeepVariant – ILMN trained | BGI-SEQ | 99.83% | 94.28% |
| DeepVariant – ILMN trained + BGI-SEQ fine-tuned | BGI-SEQ | 99.89% | 98.10% |
| DeepVariant Baseline | Illumina | 99.96% | 99.72% |
| GATK HC Baseline | Illumina | 99.87% | 98.75% |
For context, an Indel F1 (before training) of 94% would be a number you might expect from early days of GATK UnifiedGenotyper. With an Indel F1 (after training) of 98%, at the end of this tuning, DeepVariant on BGI-SEQ is about as accurate as the current version of GATK HaplotypeCaller on Illumina.
That means that in a single weekend, we could make the equivalent of several years of methods development progress. And while we don’t think that means we can take a few years off to sit on the beach, it does mean we are excited about how much further we can push this technology in the future.
Acknowledgements
We want to specifically thank the patient who donated their data for this hackathon, and whose presence and discussion at the hackathon inspired so many teams. We believe their contribution will increase the benefit of genomics for all. We thank SV.AI organizers, and all of the participants, mentors, judges and sponsors which created so much positive energy and an environment for collaboration, learning, discovery, and innovation. We also like to thank Ruibang for sharing the training data and insights for us to successfully reproduce the Clairvoyante work.



