

The DNAnexus science internship has sought to mentor students, from undergraduates to PhD students, in bioinformatics since 2015. This project was performed by Diem-Trang Pham during her internship with DNAnexus under the supervision of Sam Westreich.
We are currently recruiting summer 2019 interns. If you are interested in interning with the DNAnexus science team, please apply here.
The Problem
Investigation of microbiomes, the different types of bacteria found living together in an environment, has grown increasingly popular in recent years. With decreasing costs of sequencing and an increased number of available programs for analyzing the sequenced digital data, more researchers are turning to metagenomics – sequencing all the DNA extracted from all microbes within an environment – for examining the microbiome.
Although a number of different approaches for aligning the reads of a metagenome to a reference database have emerged over the last decade, it is tough to decide which program to use for an analysis. Each program claims to be the best option, but the authors often evaluate on their own criteria and/or test datasets. An impartial, third-party, easily replicated comparison is needed to determine how different aligners perform. This established, objective measure is currently poorly defined.
Several papers and groups have attempted to perform third-party analyses to compare different metagenome tools. Groups like CAMI hold community challenges to compare different metagenome tools. However, even in these cases, researchers must trust that the analyses were performed correctly, and there is no opportunity for researchers to update the published results with new versions of tools, different tool parameters, or different reference datasets. Individuals can download CAMI raw files for testing, but must use their own machines for processing – which can introduce even more variation. CAMI challenges and other benchmarking papers provide an important starting point, but are difficult to replicate, tough to update, and are of little use to users without access to their own computing cluster.
In order to allow for more easily replicated tests, we built new applications on the Mosaic data analytics platform. Mosaic is a community-oriented platform, powered by DNAnexus, for microbiome data analysis that allows users to create custom third-party applications. These applications run on AWS-hosted instances in the cloud, and can be run with a wide range of available hard drive space, RAM, and number of CPUs. Because users write the application code that is executed, parameters can be changed and customized for any program, and run with various combinations of RAM and available CPUs.
To ensure we accurately scored the results of different alignment tools on our metagenomes, we synthetically generated FASTQ datasets, mimicking the bacteria present in a real life gut environment. We generated low, medium, and high-complexity metagenomes based on the most abundant bacteria found within the human gut environment, using a modified version of the program metaART. Using these synthetically generated metagenomes, we ran several different aligner programs, including Bowtie2, BURST, CUSHAW3, MOSAIK, MINIMAP2, and Burrows-Wheeler Aligner (BWA). The synthetic metagenomes were compared against two references – one built from their parent genomes, and one built from 952 genomes of NCBI-identified Human microbes.
Creating Test Data
Creating a Mock Microbial Community Metagenome
We decided to build mock microbial community metagenomes to simulate gut microbiome data, in order to make confident scoring assessment of the results. We chose organisms from a list of 57 of the most common human gut microbial species, originally published by Qin et al. (2010). Qin’s paper includes both a list of species and their average relative abundances within samples. We created low, medium, and high-complexity datasets by varying the number of species included in each mock metagenome:
- The low-complexity metagenomes contain reads from the 19 most abundant species (top ⅓ of list);
- The medium-complexity metagenomes contain reads from the 28 most abundant species (top ½ of list);
- The high-complexity metagenomes contain reads from all 57 species on the list.
For each complexity level, synthetic metagenomes were produced with three different levels of quality, created by varying the insertion rate, deletion rate, and overall base quality:
| Insertion rate | Deletion rate | Base quality | |
| Low | 0.00027 – 0.00045 | 0.00033 – 0.00069 | 10 – 20 |
| Med | 0.00018 – 0.00030 | 0.00022 – 0.00046 | 10 – 30 |
| High | 0.0009 – 0.00015 | 0.00011 – 0.00023 | 10 – 40 |
 Source: https://www.nature.com/articles/nature08821
Source: https://www.nature.com/articles/nature08821
We used the program metaART to create simulated synthetic sequencing reads from our starting genomes. metaART is a wrapper for the art_illumina binary, part of the ART simulation tool. The original version of metaART was able to rapidly create simulated Illumina paired-end reads from a starting genome, but lacked the additional ability to provide custom parameters for base quality and indel rate. We made modifications to the metaART code to allow for these additional parameters to be specified and provided different values to create low, medium, and high quality synthetic metagenomes. The modified version of metaART, named “magic-metaART”, is available as an application on the Mosaic platform.
Using magic-metaART, we generated eighteen simulated metagenome datasets – nine with 100-bp paired-end reads at varying (low, medium, high) levels of quality and/or complexity, and nine with 150-bp paired-end reads at varying levels of quality and/or complexity. These simulated metagenome datasets are publicly available in Mosaic workspaces, along with the starting genomes used.
Scoring against Aligners
We aligned our simulated metagenomes of varying complexity, quality and read length against the databases of starting genomes using a variety of different alignment programs. All of these programs have been created as applications on Mosaic and are publicly available. All programs were run with default parameters; for BURST, the cutoff threshold was set at 0.97.
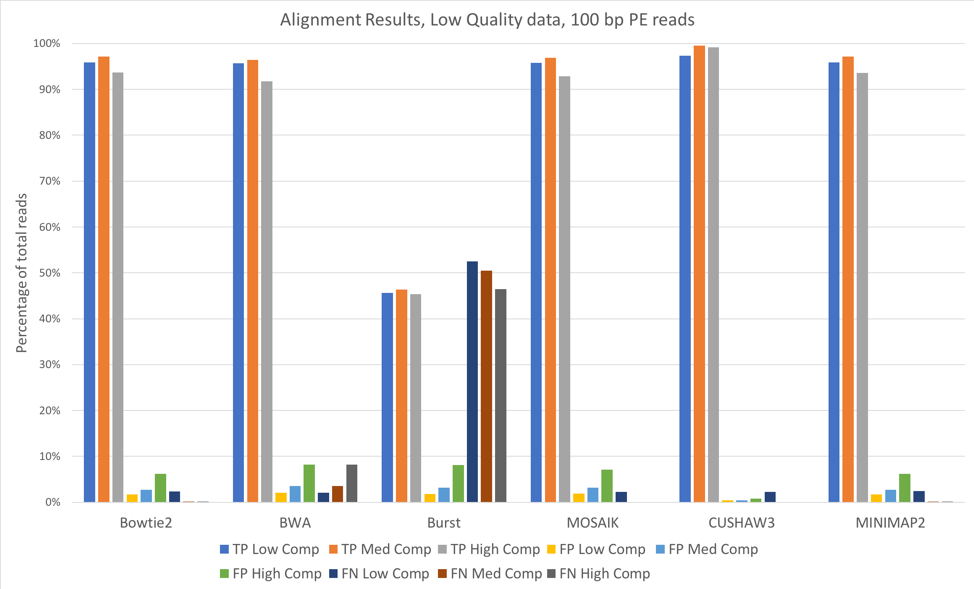
Figure 1: 100 bp reads, low quality data, aligned against known (starting) bacterial genomes
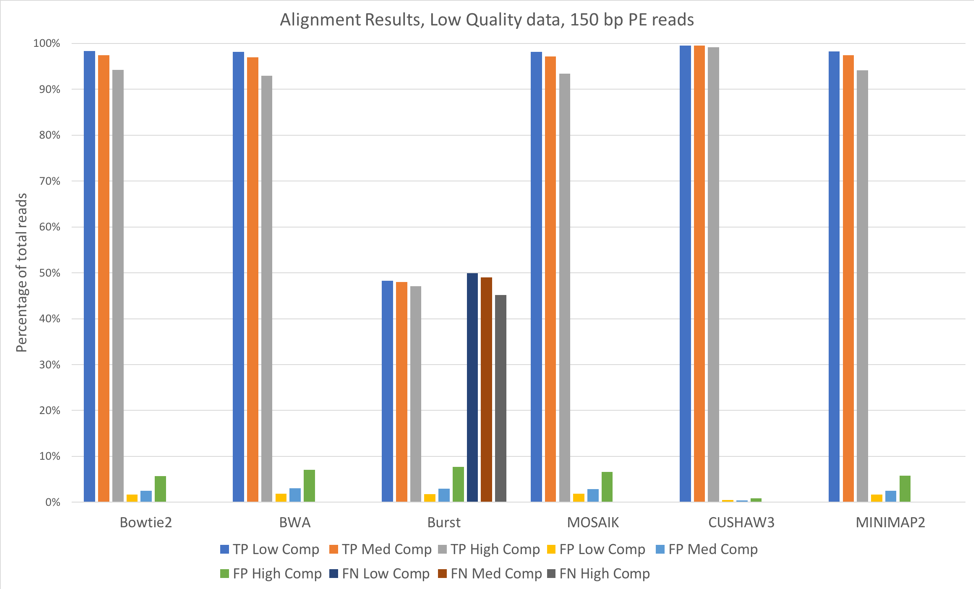
Figure 2: 150 bp reads, low quality data, aligned against known (starting) bacterial genomes
For most of these tools, the accuracy when aligning against the known bacterial genomes (the genomes used to originally construct these metagenomes) was quite high. We saw slightly better alignments when using 150 bp paired-end reads [Figure 2] when compared to 100 bp paired-end [Figure 1]. CUSHAW3 had the highest accuracy at all complexity levels, while BURST had the highest number of false negatives (reads that were not aligned to their genome of origin).
We also aligned both the 100 bp paired-end reads [Figure 3] and 150 bp paired-end reads [Figure 4] against a larger database, consisting of 952 complete genomes of known human-associated microbial species, derived from NCBI’s reference genomes collection.

Figure 3: 100 bp reads, low quality data, aligned against the NCBI 952 reference genomes
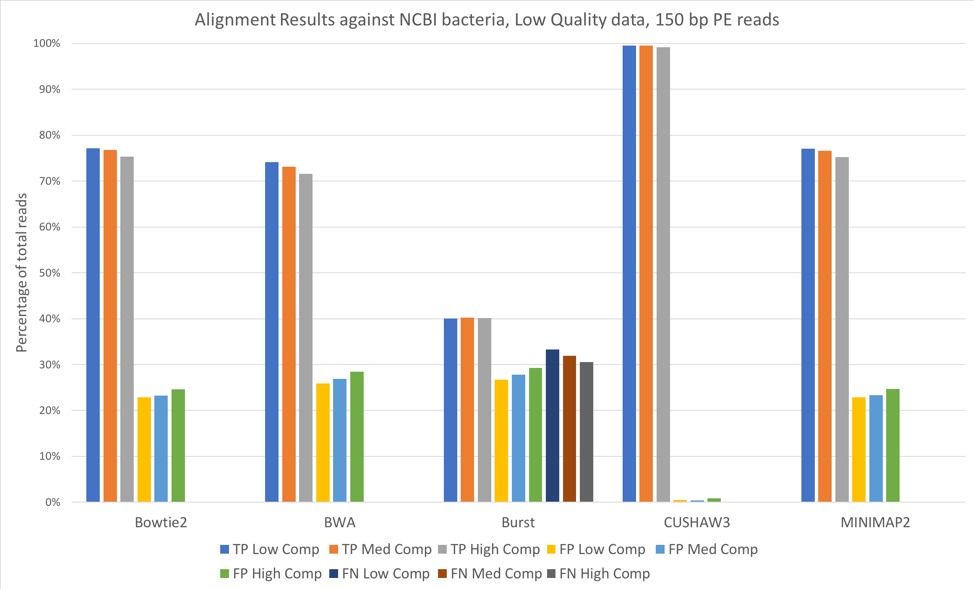
Figure 4: 150 bp reads, low quality data, aligned against the NCBI 952 reference genomes
For all tools except CUSHAW3, we observed approximately a 20-30% decrease in true positives when aligning against the larger NCBI database, with an accompanying increase in the number of false positives. Because we considered a match to any organism beyond the exact species that provided the read to be a false positive, the introduction of closely related species is likely responsible for the decreased accuracy. Once again, increased complexity led to slight decreases in the number of true positives and slight increases in the number of false positives.
Overall, we saw few performance differences between low-quality and high-quality datasets. We did note that longer reads (150 bp paired-end vs. 100 bp paired-end) increased the number of true positives. We also observed that increases in complexity led to slight decreases in the number of true positives. Against both reference databases, CUSHAW3 correctly aligned the highest percentage of reads, while BURST had a higher number of false negatives than the other aligners.
Evaluating the Resource Requirements of Aligners
We observed the amount of time and memory needed to run each aligner for two steps: indexing the reference, and aligning metagenomic reads against the reference. Building an index is often more intensive, due to the amount of data that must be read and processed, although this usually only needs to be done once. After a reference is indexed, it can be used for an unlimited number of searches.
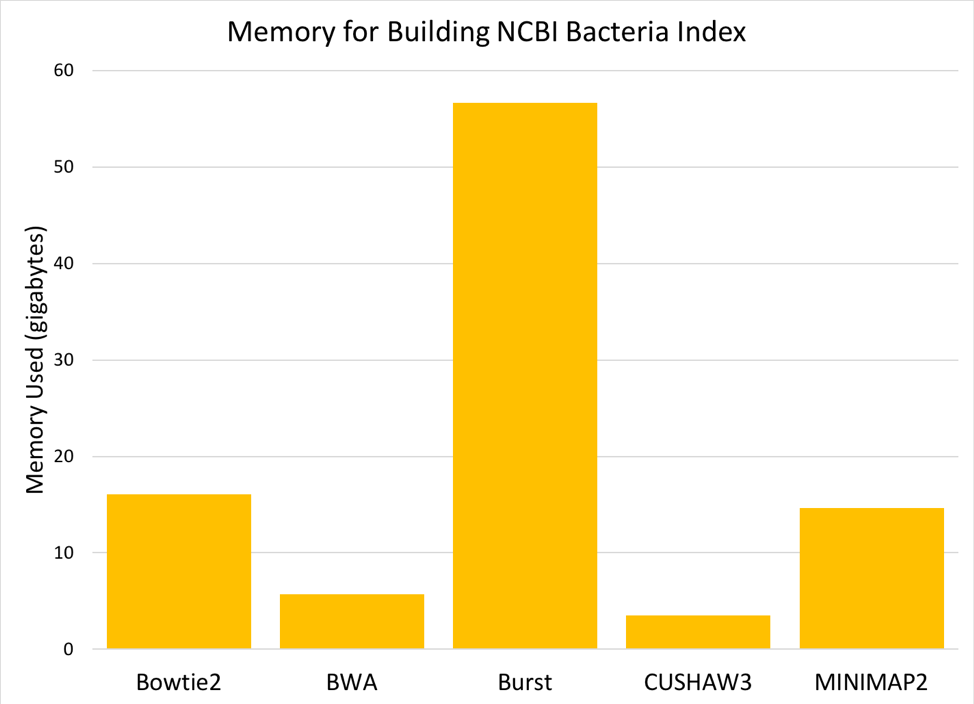
Figure 5: Memory (gigabytes) needed to build a searchable index from the 952 NCBI reference genomes.
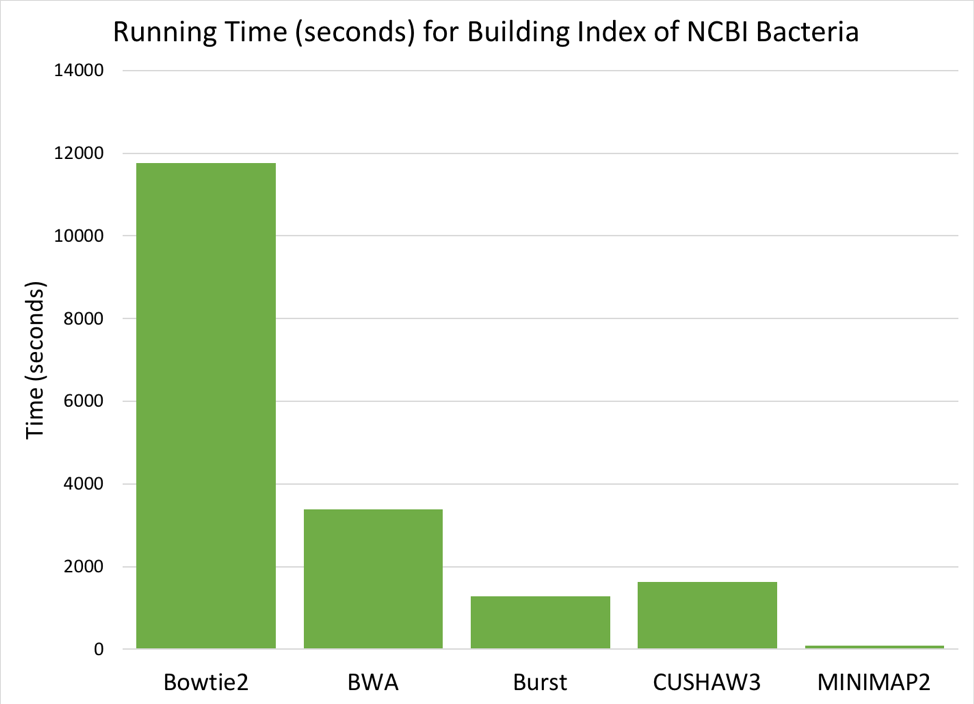
Figure 6: Time (seconds) needed to build a searchable index from the 952 NCBI reference genomes.
Comparing the six aligners, we saw large differences in memory and time required [Figures 5 & 6]. MOSAIK is not displayed in the graphs, as it failed to successfully finish building an index before the instance timed out (>24 hours). BURST requires the most memory to build an index, while BWA and CUSHAW3 both have very low memory requirements. Of the aligners that successfully built an index, BOWTIE2 was much slower than the others, while MINIMAP2 was fastest by a significant margin.
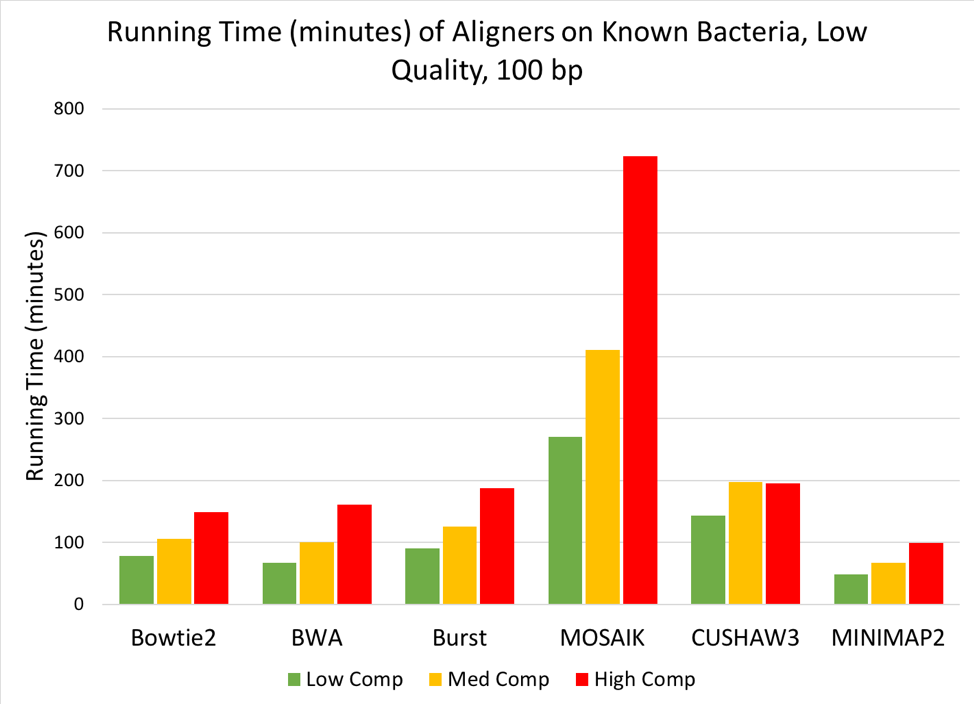
Figure 7: Time (minutes) needed to align low, medium, or high complexity simulated metagenomes (100bp, low quality reads) against the Known Bacteria reference.
When aligning the simulated metagenomes against each reference, we saw a linear increase in run time as the complexity of the metagenome increased [Figure 7], but no change in run time due to the quality of the simulated metagenome. When aligning against the index of known bacteria used to generate the simulated metagenomes, MOSAIK required the longest time to align reads to the reference, while all other tools finished the alignments in under 200 minutes. MINIMAP2 performed the fastest of the 6 tools, finishing even the high-complexity metagenome in under two hours.
Relative Tool Performance versus Cost
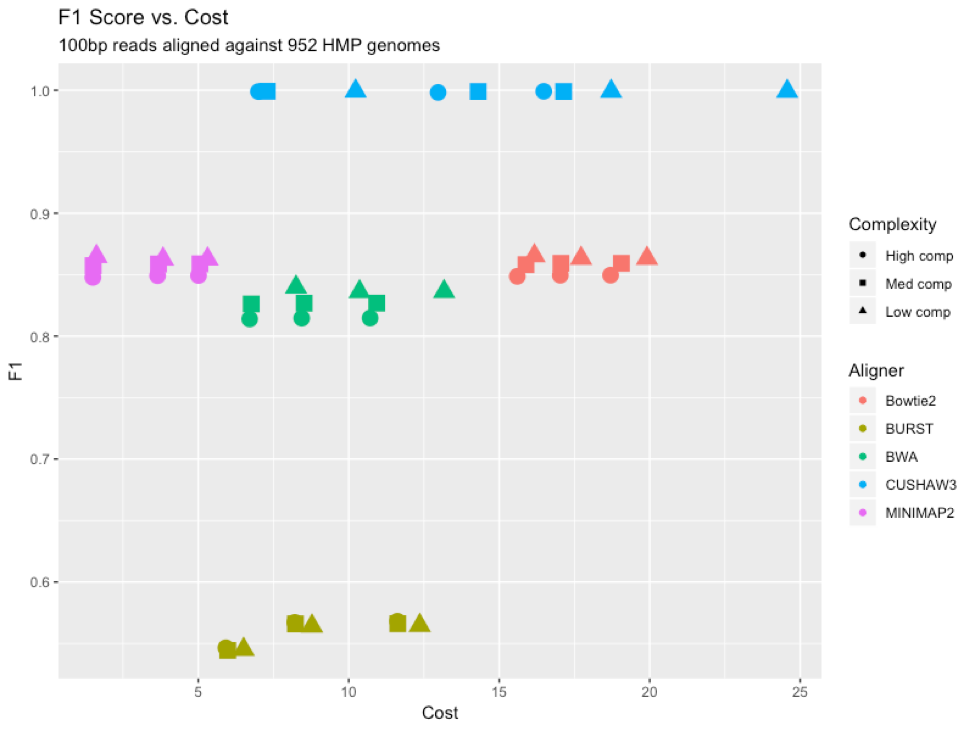
Figure 8: F1 scores for each aligner (color) at each complexity level (shape), graphed versus cost (dollars) to run each program on the Mosaic platform.
To provide an “overall” assessment of the performance of the different aligners, we looked at the F1 score (the harmonic average of precision and recall) for each tool compared to its cost to run on the Mosaic platform [Figure 8]. CUSHAW3 had the highest F1 score, but also proved to be the most expensive to run, due largely to its long run time for high-complexity simulated metagenomes. BURST had the lowest F1 score, lagging behind the other aligners due to an increased number of false negatives. MINIMAP2 proved to be the cheapest aligner to run, due to its rapid time and reasonable memory demands.
Abundance Profile Accuracy After Alignment
Although the percentage of aligned reads is a useful observation, the true question is how the results of each aligner predicts the actual abundance profile from the simulated metagenomes. In other words, how well do the calculated abundance profiles match the original abundances of the genomes used to create the simulated data?
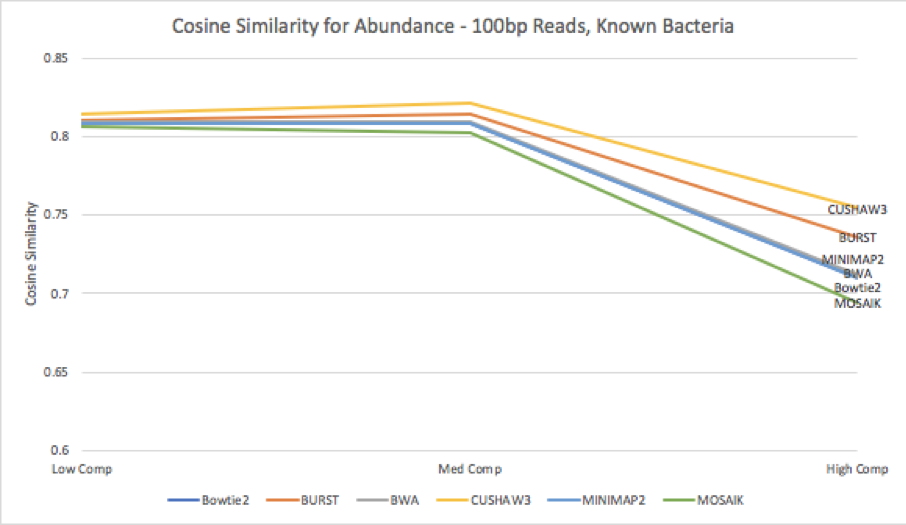
Figure 9: Cosine similarity for abundance profiles calculated from each aligner’s results, run against the known bacteria reference.
When we compare the estimated abundances against the original abundances from the results matched against the database of known bacteria [Figure 9], we see that the variance increases for all tools as complexity increases. This increase in variance leads to a decrease in cosine similarity for all tools; CUSHAW3’s shows the least decrease, while MOSAIK’s results are the most dissimilar from the truth. Surprisingly, although BURST had the highest number of discarded reads, it appears to still provide a fairly accurate estimate of the abundances of different organisms within the simulated metagenome.

Figure 10: Cosine similarity for abundance profiles calculated from each aligner’s results, run against the NCBI bacteria reference.
When we examine the estimated abundances from alignment against the NCBI database of bacteria [Figure 10], we see an interesting story; MINIMAP2, BWA, and Bowtie2 all perform at a very similar level, while BURST and CUSHAW3 lag slightly behind. In contrast to the abundance estimates against solely known bacteria, these results increase in accuracy as the complexity of the simulated metagenome increases; we hypothesize that some of the reads always map to incorrect annotations, and thus adding more diversity reduces their negative impact on cosine similarity score.
Conclusions
Although it is extremely difficult to conclude that any one alignment tool is objectively better than another, we have showed that, through the use of Mosaic applications, we can evaluate the performance of multiple alignment tools on simulated metagenomic datasets. We created simulated metagenomic datasets of varying quality and complexity, modeled after the most abundant organisms found within the gut microbiome. We aligned these simulated metagenomes against two references of different sizes and observed the accuracy, memory requirement, and time requirement of each program.
Overall, we observed the highest alignment accuracy from CUSHAW3, and the highest speed from MINIMAP2. Bowtie2 and BWA both lagged in accuracy when compared to CUSHAW3, and ran slower than MINIMAP2 without offering any additional accuracy. BURST had the highest number of false negatives in alignments.
When we examined how the number of annotations impacted estimated abundance, we found that, although BURST drops significantly more reads than the other aligners, these dropped reads do not cause an overly large decrease in abundance accuracy estimation compared to other tools.
In conclusion, we show that different aligner tools have varying performances in regard to accuracy, speed, and memory usage. We strongly encourage that, when developing an alignment-based pipeline for metagenomic data, users test multiple aligners to compare the results for optimal speed and accuracy.
Program and File Resources
Availability of Mock Metagenomes
The 100bp simulated gut metagenomes are available here.
The 150bp simulated gut metagenomes are available here.
All of the alignment tools are publicly available to run on Mosaic.
Tool versions: ART – 2.5.8; Bowtie2 – 2.3.4.1; BWA – 0.7.17; BURST – 0.99.7; MOSAIK – 2.2.26; CUSHAW3 – 3.0.3; SAMtools – 1.9; MINIMAP2 – 2.12
Reference
Qin et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing.




