

Researchers from academia and industry, including Li Lab (Dana-Farber Cancer Institute), Church Lab (Wyss Institute, Harvard University), DNAnexus Research Lab, and others, have developed a new genome assembly approach , dubbed DipAsm, for generating chromosome-scaled phased contigs using long reads and long-range confirmation data. The method, described in Nature Biotechnology, could generate results within a day and outperforms other approaches in terms of contiguity and completeness of the phased assemblies. As shown in the paper, when the method was applied to four public datasets, it produced haplotype-resolved assemblies with contig NG50 of up to 25 Mb and phased almost all heterozygous sites with 98-99 percent accuracy.
Being able to generate accurate chromosome-scale haplotype-resolved assemblies is crucial for capturing the heterozygous variation present in human genomes and understanding allele-specific methylation and gene expression in research and clinical applications. The accuracy of DipAsm’s assemblies makes it a valuable tool for exploring highly polymorphic parts of the genome such as the Human Leukocyte Antigen region, and the Killer-cell Immunoglobulin-like Receptor region. “Our phased assemblies can reconstruct most of these regions with two contigs for each haplotype,” first author Shilpa Garg, a postdoctoral researcher in the Li Lab, explained in the paper. They also demonstrate how the method enables the identification of both SNPs and structural variants with greater sensitivity and specificity than some current methods.
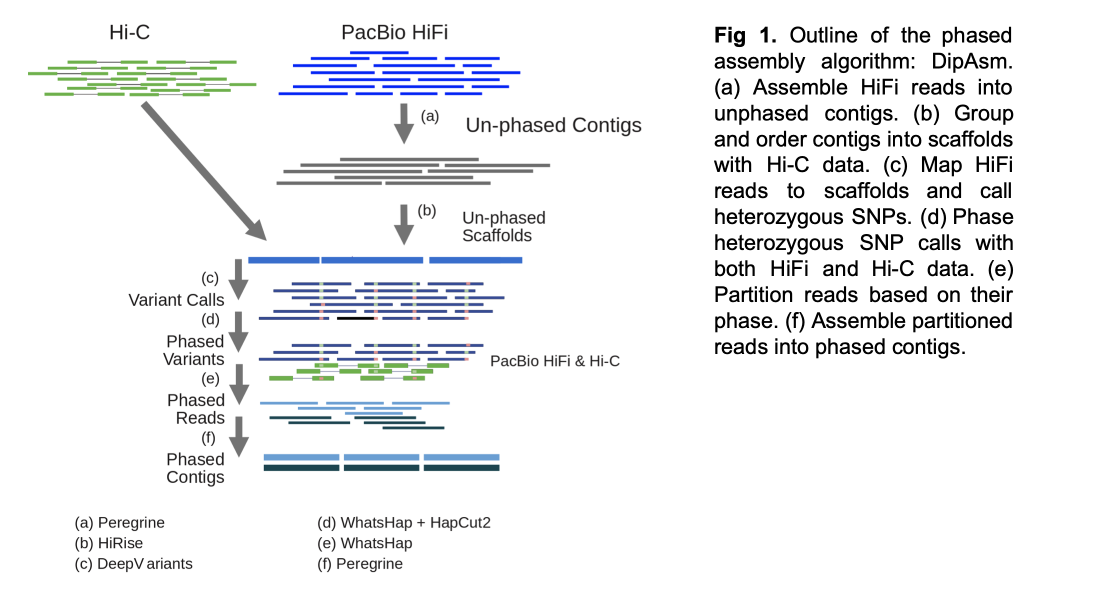
Full details of how DipAsm works are provided in the paper. But briefly, it reconstructs the haplotypes present in diploid individuals using PacBio’s long High-Fidelity reads and Hi-C data as input. DipAsm works with data from an unphased Peregrine assembly scaffolded by 3D-DNA or HiRise. It uses DeepVariant to call small variants, phases them using WhatsHap and HapCUT2, partitions the reads, and then assembles each partition independently using the Peregrine assembly toolkit.
To demonstrate the method’s accuracy, the researchers applied it to data from four human genomes: the PGP1 from the Personal Genome Project, HG002 and NA12878 from the Genome In a Bottle dataset (GIAB), and HG00733 from the HGSVC project. Full details of the assembly statistics including specifics about the assemblies used for the comparison to the results of the DipAsm pipeline are provided in Table 1 in the paper.
From the GIAB HG002 sample, the researchers generated a phased de novo assembly of 5.95 gigabases that incorporated data from both parental haplotypes. Compared to results from trio binning-based assemblies, the DipAsm assembly achieved better contiguity, and disagreed with less than 0.5% of phased heterozygous SNPs.
To evaluate the consensus accuracy of the DipAsm assembly, the researchers used dipcall to align the phased contigs of the HG002 that they created against the human reference genome. Next, they called SNPs and insertions and deletions from the alignment and compared these calls to the GIAB truth dataset. Out of the 2.36Gb confident regions in GIAB, the DipAsm assembly generated over 5,700 false SNP alleles (about 0.19% of called SNPs), and over 65,000 false insertion and deletion alleles (over 11% of called indels). It “achieves a consensus accuracy comparable to the Arrow-polished TrioCanu assembly,” according to the researchers.
Comparing the assembly to the GIAB truth data demonstrates DipAsm’s phasing power. “During assembly, failing to partition reads in heterozygous regions leads to the loss of heterozygotes,” the team explains. “On this metric, our Hi-C based assemblies only miss 0.4% of heterozygous SNPs. Those results are about 8 times better than those gleaned from a trio binning-based assembly, which is less powerful potentially because it is unable to phase a heterozygote when all individuals in a trio are heterozygous at the same site,” the researchers noted in the paper. Furthermore, “trio binning breaks short reads into k-mers, which also reduces power in comparison to mapping full-length paired-end Hi-C reads in our pipeline.”
In terms of long indels (>50bp), the DipAsm assembly-based call set showed over 93% sensitivity and 92% precision compared to the GIAB structural variant truth dataset. In comparison, trio binning-based call sets had about 3% lower sensitivity for indels and small variants. The researchers also identified various structural variants in the DipAsm haplotype assemblies including microsatellites, simple repeats, and short interspersed nuclear elements.
Other results reported in the paper describe findings from comparing phased SNP calls from the DipAsm version of the HG00733 assembly to calls from the Human Genome Structural Variation Consortium. The results showed that the DipAsm assembly had a slightly lower phasing error rate and phased more heterozygous SNPs. The team also used DipAsm to assemble the NA12878 and PGP1 genomes. And those results showed that “we can achieve chromosome-long phasing albeit the shorter read length of NA12878 and the lower read coverage of PGP1,” the researchers wrote. Comparisons of these assemblies to those in the GIAB truth set indicated that DipAsm’s NA12878 assembly offers better consensus accuracy.




