

Although we’ll miss the chance to spend some time in San Diego, we’re excited for the opportunity to expose our research to the wider world, as the popular annual meeting of the American Society of Human Genetics (ASHG) goes virtual this year.
From pharmacogenomics to frameworks for benchmarking, the DNAnexus research team will be presenting posters on a number of exciting topics. We’ve included them all below, for easy reference.
Are you susceptible to adverse drug reactions?

Pharmacogenomics (PGx) is an embodiment of precision medicine. Yet gene-drug specific dosing guidelines are still limited. Using LightGBM – a decision-tree based gradient boosting machine learning framework – and UK Biobank phenotypic and genomic data, Chiao-Feng Lin and colleagues from the xVantage and Apollo Data Science teams investigated whether it is possible to predict an individual’s risk to adverse drug reactions, irrespective of drugs. Check out her Reviewers Choice Award poster to learn what they found, and how the Apollo Cohort Browser can make sample selection simpler.
Poster 3591
The Portable Workflow Environs: Cloud-scale workflows with nano-scale effort
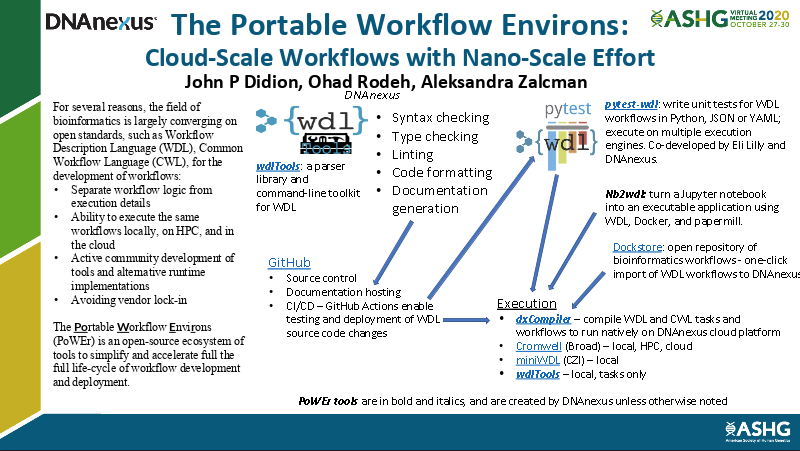
As the field of bioinformatics is largely converging on open standards, such as Workflow Description Language (WDL), for the development of workflows, we have developed a suite of tools that can be leveraged for their efficient and scalable development and deployment. Join John Didion as he introduces PoWEr (The Portable Workflow Environs), an open-source ecosystem of tools to simplify and accelerate the full life-cycle of workflow development and deployment.
Poster 2217
Direct-to-risk: A scalable framework for end-to-end GWAS, fine-mapping and risk prediction on UK Biobank

While polygenic risk score (PGS) analyses seem straightforward, they consist of a plethora of disparate steps, each with their own datasets, methods, and hidden assumptions. DNAnexus Apollo can be used to conduct end-to-end PGS analyses with semi-automation, scalability, traceability, interoperability, and iterability. Peter Nguyen will demonstrate how, using the framework to conduct PGS analyses for type 2 diabetes by processing phenotypic and genomic data across 500,000 thousand UK Biobank participants and 90 million variants.
Poster 3802
A WDL-based framework for benchmarking germline variant calling pipelines for high-throughput sequencing data
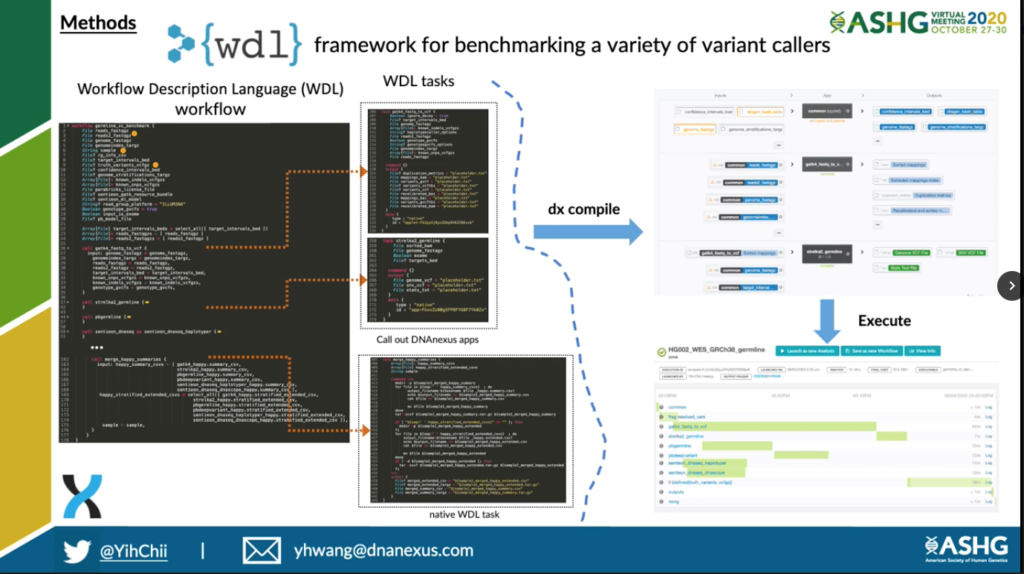
Wondering which variant caller would work best for you? We developed a WDL-based framework and benchmarked six variant callers for their accuracy and runtime. Join Yih-Chii Hwang as she describes the flexible framework and how it can be further customized and applied to benchmark any bioinformatics pipelines of interest.
Poster 2018
In addition to our own research, we were proud to contribute to the following work by our customers and partners:
A new Genome in a Bottle benchmark for hard to assess and medically important genes

We were proud to contribute to this project, whose goal was to create a high quality benchmark variant call set for medically relevant genes, to help researchers and clinical related applications with the next gene sequencing. Be sure to check out the Reviewers Choice Award poster by the multi-institutional Genome In a Bottle team.
Poster 2009
Automated repeat characterization of filaggrin from PacBio Sequel HiFi long reads
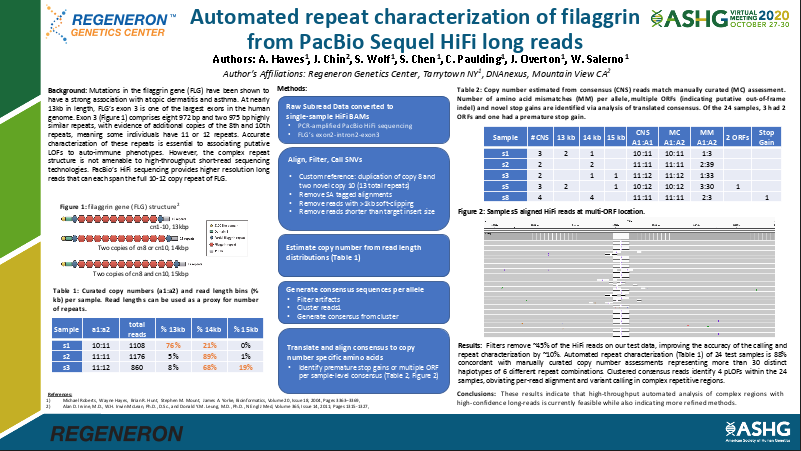
Filaggrin (FLG) is a medically important protein coding gene, associated with dermatitis, atopic, ichthyosis vulgaris, and other conditions. This poster demonstrates how to do efficient variant calling for this repeat-rich gene using a new method based on PacBio SMRT Sequencing. A DNAnexus applet was also created to help our collaborators at the Regeneron Genetics Center process thousands of samples for their research work on the related diseases.
Poster 2046
Doubly Confused: Evaluation of splicing variant impact assessment with computational prediction, and vice versa
Many variants affecting splicing remain of unknown significance in the absence of definitive molecular and/or clinical data. Using data from three carefully chosen genes — HPRT, BRCA1, and ABCA4 — we helped scientists at the University of Maryland assess the quality of splice variant impact assessment software. Check out the poster to see which software came ahead.
Poster 2088
Sequencing Quality Control Phase II: Inter and intra variability of NGS and its implication on structural variation detection
Genomic structural variation (SV) includes several different classes of mutations, including deletions, insertions, translocations, duplications, inversions and complex rearrangements. The complexity of these different events continues to make the detection of SV challenging. This is amplified by the usage of different sequencing technologies, centers, or across replicates within and between cohort studies. To better understand the impact of variability due to preparation, sequencing and analysis, we helped Baylor scientists compare the sequence of DNA from a Chinese family consisting of two identical twins and their parents at three different sequencing centers with three replicates each as part of the Sequencing Quality control Phase II (SEQC2) study. We utilized multiple analytical pipelines and compared the resulting 288 data sets within and across the family members. Check out the poster to see what was found.
Poster 2202



